
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- My Bibliography
- Collections
- Citation manager

Save citation to file
Email citation, add to collections.
- Create a new collection
- Add to an existing collection
Add to My Bibliography
Your saved search, create a file for external citation management software, your rss feed.
- Search in PubMed
- Search in NLM Catalog
- Add to Search
Prodrugs in medicinal chemistry and enzyme prodrug therapies
Affiliations.
- 1 Department of Chemistry, Aarhus University, Denmark.
- 2 School of Pharmacy, University of Eastern Finland, Kuopio, Finland. Electronic address: [email protected].
- 3 Department of Chemistry, Aarhus University, Denmark; iNano Interdisciplinary Nanoscience Centre, Aarhus University, Denmark. Electronic address: [email protected].
- PMID: 28676386
- DOI: 10.1016/j.addr.2017.06.013
Prodrugs are cunning derivatives of therapeutic agents designed to improve the pharmacokinetics profile of the drug. Within a prodrug, pharmacological activity of the drug is masked and is recovered within the human body upon bioconversion of the prodrug, a process that is typically mediated by enzymes. This concept is highly successful and a significant fraction of marketed therapeutic formulations is based on prodrugs. An advanced subset of prodrugs can be engineered such as to achieve site-specific bioconversion of the prodrug - to comprise the highly advantageous "enzyme prodrug therapy", EPT. Design of prodrugs for EPT is similar to the prodrugs in general medicinal use in that the pharmacological activity of the drug is masked, but differs significantly in that site-specific bioconversion is a prime consideration, and the enzymes typically used for EPT are non-mammalian and/or with low systemic abundance in the human body. This review focuses on the design of prodrugs for EPT in terms of the choice of an enzyme and the corresponding prodrug for bioconversion. We also discuss the recent success of "self immolative linkers" which significantly empower and diversify the prodrug design, and present methodologies for the design of prodrugs with extended blood residence time. The review aims to be of specific interest for medicinal chemists, biomedical engineers, and pharmaceutical scientists.
Keywords: Drug delivery; Enzyme; Enzyme prodrug therapy; Prodrugs; Self immolative linkers.
Copyright © 2017 Elsevier B.V. All rights reserved.
PubMed Disclaimer
Similar articles
- Substrate mediated enzyme prodrug therapy. Fejerskov B, Jarlstad Olesen MT, Zelikin AN. Fejerskov B, et al. Adv Drug Deliv Rev. 2017 Sep 1;118:24-34. doi: 10.1016/j.addr.2017.04.013. Epub 2017 Apr 27. Adv Drug Deliv Rev. 2017. PMID: 28457884 Review.
- Prodrug applications for targeted cancer therapy. Giang I, Boland EL, Poon GM. Giang I, et al. AAPS J. 2014 Sep;16(5):899-913. doi: 10.1208/s12248-014-9638-z. Epub 2014 Jul 9. AAPS J. 2014. PMID: 25004822 Free PMC article. Review.
- Designing metal-contained enzyme mimics for prodrug activation. Du B, Li D, Wang J, Wang E. Du B, et al. Adv Drug Deliv Rev. 2017 Sep 1;118:78-93. doi: 10.1016/j.addr.2017.04.002. Epub 2017 Apr 12. Adv Drug Deliv Rev. 2017. PMID: 28412325 Review.
- Self-immolative nitrogen mustard prodrugs for suicide gene therapy. Niculescu-Duvaz D, Niculescu-Duvaz I, Friedlos F, Martin J, Spooner R, Davies L, Marais R, Springer CJ. Niculescu-Duvaz D, et al. J Med Chem. 1998 Dec 17;41(26):5297-309. doi: 10.1021/jm980425k. J Med Chem. 1998. PMID: 9857097
- Self-Immolative Linkers Literally Bridge Disulfide Chemistry and the Realm of Thiol-Free Drugs. Riber CF, Smith AA, Zelikin AN. Riber CF, et al. Adv Healthc Mater. 2015 Aug 26;4(12):1887-90. doi: 10.1002/adhm.201500344. Epub 2015 Jun 24. Adv Healthc Mater. 2015. PMID: 26109168
- A Review on Carbon Nanotubes Family of Nanomaterials and Their Health Field. Brito CL, Silva JV, Gonzaga RV, La-Scalea MA, Giarolla J, Ferreira EI. Brito CL, et al. ACS Omega. 2024 Feb 13;9(8):8687-8708. doi: 10.1021/acsomega.3c08824. eCollection 2024 Feb 27. ACS Omega. 2024. PMID: 38434894 Free PMC article. Review.
- Polarization of macrophages to an anti-cancer phenotype through in situ uncaging of a TLR 7/8 agonist using bioorthogonal nanozymes. Zhang X, Liu Y, Jiang M, Mas-Rosario JA, Fedeli S, Cao-Milan R, Liu L, Winters KJ, Hirschbiegel CM, Nabawy A, Huang R, Farkas ME, Rotello VM. Zhang X, et al. Chem Sci. 2024 Jan 9;15(7):2486-2494. doi: 10.1039/d3sc06431j. eCollection 2024 Feb 14. Chem Sci. 2024. PMID: 38362405 Free PMC article.
- Advance Progress in Assembly Mechanisms of Carrier-Free Nanodrugs for Cancer Treatment. Zhang X, Hu S, Huang L, Chen X, Wang X, Fu YN, Sun H, Li G, Wang X. Zhang X, et al. Molecules. 2023 Oct 13;28(20):7065. doi: 10.3390/molecules28207065. Molecules. 2023. PMID: 37894544 Free PMC article. Review.
- Novel drug delivery strategies for antidepressant active ingredients from natural medicinal plants: the state of the art. Yuan S, Ma T, Zhang YN, Wang N, Baloch Z, Ma K. Yuan S, et al. J Nanobiotechnology. 2023 Oct 27;21(1):391. doi: 10.1186/s12951-023-02159-9. J Nanobiotechnology. 2023. PMID: 37884969 Free PMC article. Review.
- Ce-based solid-phase catalysts for phosphate hydrolysis as new tools for next-generation nanoarchitectonics. Komiyama M. Komiyama M. Sci Technol Adv Mater. 2023 Sep 8;24(1):2250705. doi: 10.1080/14686996.2023.2250705. eCollection 2023. Sci Technol Adv Mater. 2023. PMID: 37701758 Free PMC article.
Publication types
- Search in MeSH
LinkOut - more resources
Full text sources.
- Elsevier Science
- Ovid Technologies, Inc.
Other Literature Sources
- The Lens - Patent Citations
- scite Smart Citations
- Citation Manager
NCBI Literature Resources
MeSH PMC Bookshelf Disclaimer
The PubMed wordmark and PubMed logo are registered trademarks of the U.S. Department of Health and Human Services (HHS). Unauthorized use of these marks is strictly prohibited.

Challenges and Rewards
- © 2007
- Valentino J. Stella 0 ,
- Ronald T. Borchardt 1 ,
- Michael J. Hageman 2 ,
- Reza Oliyai 3 ,
- Hans Maag 4 ,
- Jefferson W. Tilley 5
University of Kansas, Lawrence, USA
You can also search for this editor in PubMed Google Scholar
Bristol-Myers Squibb Company, Princeton, USA
Gilead sciences, foster city, usa, roche palo alto, llc, palo alto, usa, hoffmann-la roche, inc., nutley, usa.
- Represents the first text of its kind, designed to give the reader a complete overview of prodrugs: the current status of the prodrug concept and its many applications and successes in overcoming formulation and delivery of problematic drugs
- Replete with examples of approved and marketed prodrugs, including 25 case studies of marketed prodrugs from which one might learn and apply to future strategic endeavors in the development of prodrug candidates
- Introduces the topic to the novice as well as professional in the design of prodrugs
Part of the book series: Biotechnology: Pharmaceutical Aspects (PHARMASP, volume V)
422k Accesses
322 Citations
16 Altmetric
This is a preview of subscription content, log in via an institution to check access.
Access this book
Subscribe and save.
- Get 10 units per month
- Download Article/Chapter or eBook
- 1 Unit = 1 Article or 1 Chapter
- Cancel anytime
- Available as PDF
- Read on any device
- Instant download
- Own it forever
- Compact, lightweight edition
- Dispatched in 3 to 5 business days
- Free shipping worldwide - see info
- Durable hardcover edition
Tax calculation will be finalised at checkout
Other ways to access
Licence this eBook for your library
Institutional subscriptions
About this book
Similar content being viewed by others.

The expanding role of prodrugs in contemporary drug design and development

The landscape of small-molecule prodrugs

Prodrug Design to Enhance Bioavailability and Systemic Delivery
Table of contents (57 chapters), front matter, a case for prodrugs.
- Valentino J. Stella
Problems Addressable by Prodrugs
Permeability, prodrug approaches to enhancing the oral delivery of poorly permeable drugs, topical delivery using prodrugs.
- Kenneth B. Sloan, Scott C. Wasdo
Prodrug approaches to ophthalmic drug delivery
- Tomi Järvinen, Riku Niemi
Overcoming Poor Aqueous Solubility of Drugs for Oral Delivery
- Tycho Heimbach, David Fleisher, Amal Kaddoumi
Prodrugs and Parenteral Drug Delivery
- Jeffery Hemenway, Valentino J. Stella
Poly (ethylene glycol) Prodrugs: Altered Pharmacokinetics and Pharmacodynamics
- Richard B. Greenwald, Hong Zhao
Prodrugs to Reduce Presystemic Metabolism
- Bruce J. Aungst, Nicole Matz
Controlled Release
Controlled release — small molecules.
- Jaymin C. Shah
Controlled Release - Macromolecular Prodrugs
- Claus Larsen, Jesper Østergaard, Susan W. Larsen
Controlled Release - Proenzymes
- Richard L. Schowen
Targeting - Theoretical and Computational Models
- Roger A. Rajewski, Michelle P. McIntosh
Targeting - Cancer — Small Molecules
- Vincent (F.M.H.) de Groot
Monoclonal Antibody Drug Conjugates for Cancer Therapy
- Peter D. Senter, Damon L. Meyer
Antibody-Directed Enzyme Prodrug Therapy
- Kenneth D. Bagshawe
Prodrugs for Liver-targeted Drug Delivery
- Mark D. Erion
Prodrug Approaches for Drug Delivery to the Brain
- Bradley D. Anderson
Editors and Affiliations
Valentino J. Stella, Ronald T. Borchardt
Michael J. Hageman
Reza Oliyai
Jefferson W. Tilley
Bibliographic Information
Book Title : Prodrugs
Book Subtitle : Challenges and Rewards
Editors : Valentino J. Stella, Ronald T. Borchardt, Michael J. Hageman, Reza Oliyai, Hans Maag, Jefferson W. Tilley
Series Title : Biotechnology: Pharmaceutical Aspects
DOI : https://doi.org/10.1007/978-0-387-49785-3
Publisher : Springer New York, NY
eBook Packages : Biomedical and Life Sciences , Biomedical and Life Sciences (R0)
Copyright Information : Springer-Verlag New York 2007
Hardcover ISBN : 978-0-387-49782-2 Published: 12 March 2007
Softcover ISBN : 978-1-4939-5050-8 Published: 04 May 2017
eBook ISBN : 978-0-387-49785-3 Published: 26 August 2007
Edition Number : 1
Number of Pages : XVIII, 1464
Topics : Pharmacology/Toxicology
- Publish with us
Policies and ethics
Societies and partnerships

- Find a journal
- Track your research
Information
- Author Services
Initiatives
You are accessing a machine-readable page. In order to be human-readable, please install an RSS reader.
All articles published by MDPI are made immediately available worldwide under an open access license. No special permission is required to reuse all or part of the article published by MDPI, including figures and tables. For articles published under an open access Creative Common CC BY license, any part of the article may be reused without permission provided that the original article is clearly cited. For more information, please refer to https://www.mdpi.com/openaccess .
Feature papers represent the most advanced research with significant potential for high impact in the field. A Feature Paper should be a substantial original Article that involves several techniques or approaches, provides an outlook for future research directions and describes possible research applications.
Feature papers are submitted upon individual invitation or recommendation by the scientific editors and must receive positive feedback from the reviewers.
Editor’s Choice articles are based on recommendations by the scientific editors of MDPI journals from around the world. Editors select a small number of articles recently published in the journal that they believe will be particularly interesting to readers, or important in the respective research area. The aim is to provide a snapshot of some of the most exciting work published in the various research areas of the journal.
Original Submission Date Received: .
- Active Journals
- Find a Journal
- Proceedings Series
- For Authors
- For Reviewers
- For Editors
- For Librarians
- For Publishers
- For Societies
- For Conference Organizers
- Open Access Policy
- Institutional Open Access Program
- Special Issues Guidelines
- Editorial Process
- Research and Publication Ethics
- Article Processing Charges
- Testimonials
- Preprints.org
- SciProfiles
- Encyclopedia
Article Menu
- Subscribe SciFeed
- Recommended Articles
- PubMed/Medline
- Google Scholar
- on Google Scholar
- Table of Contents
Find support for a specific problem in the support section of our website.
Please let us know what you think of our products and services.
Visit our dedicated information section to learn more about MDPI.
JSmol Viewer
Prodrugs of nonsteroidal anti-inflammatory drugs (nsaids), more than meets the eye: a critical review.

1. Introduction
1.1. prodrugs, 1.2. non-steroidal anti-inflammatory drugs (nsaids), 1.3. nsaid prodrugs, 2. no-nsaids, 2.1. introduction, 2.2. no-nsaids with intrinsic pharmacological activity.
- A group of glyceryl dinitrate esters ( 1a – c ) and NONOate-containing prodrugs ( 2a – c ) of aspirin ( a = ASA ), indomethacin ( b = IND ) and ibuprofen ( c = IBU ) ( Figure 7 ) have been synthesized and evaluated in vivo and in vitro for their anti-inflammatory activity [ 52 ]. Although the ibuprofen and indomethacin esters showed in vivo activity comparable to their parent NSAIDs, the aspirin esters showed less than half the activity of aspirin. Furthermore, none of them showed in vitro inhibitory activity against COX-1; rather, they all showed inhibitory activity against COX-2 ( in vitro IC 50 = 0.6–9.3 μM) that was comparable to or greater than that of their parent NSAIDs. These results pose two questions: was the in vivo biological activity of these compounds due solely to the parent NSAIDs? And, second, was the GI-sparing profile only the result of NO release?
- The ethanesulfohydroxamic acid esters of indomethacin ( 3 ) and naproxen ( 4 ) were synthesized and evaluated as NO-NSAIDs [ 47 ] ( Figure 8 ). The indomethacin ester 3 was a selective COX-2 inhibitor (IC 50 = 0.42 μM) in vitro , with an in vivo ID 50 of 19.1 μmol/kg compared to 11.7 μmol/kg for indomethacin. Furthermore, the non-NO-releasing hydroxamic acid ester of ibuprofen 5 showed comparable in vivo potency (78.9% inhibition of inflammation at 327 μmol/kg oral dose) to its NO-releasing hydroxamic acid (79.5% inhibition of inflammation at 327 μmol/kg oral dose). It is worth mentioning that the IC 50 of 5 against COX-2 was 0.63 μM. This adds more skepticism to the accepted notion(s) that NO-releasing esters of NSAIDs are mere prodrugs and/or that NO release is essential for their GI-sparing profile. The possibility that compound 5 is a prodrug still exists, but no investigation of this possibility was performed.
- The ester of diclofenac ( 6 ) ( Figure 9 ) contains benzofuroxan as the NO-releasing group. Ester 6 was synthesized and evaluated in vitro and in vivo for the reduction of PGE 2 and TXB 2 levels in plasma. This ester was found to be more effective in inhibiting PGE 2 synthesis than TXB 2 synthesis in vitro . This finding suggests that this NO-diclofenac ester is more selective for COX-2 [ 46 ].
- 2-Hydroxysulfamoylbenzoic acid 7 and its ethyl benzoate ester 8 ( Figure 10 ), which are nitric oxide-releasing analogs of aspirin, were reported by Kaur et al. (2012) [ 53 ]. Although not prodrugs, these compounds show that the simple backbone of aspirin, a nonselective COX inhibitor, can be modified to become a selective COX-2 inhibitor [ 53 ]. The pharmacological evaluation of compounds 7 and 8 revealed that hydroxamic acid 8 is as a potent and a much more selective COX-2 inhibitor than celecoxib (IC 50 = 0.09 μM, with more than 1000-fold selectivity). In addition, hydroxamic acid 8 was found to be a 5-lipoxygenase (5-LOX) inhibitor (IC 50 = 0.4 μM). 5-LOX is an essential enzyme in the biosynthetic pathway of leukotrienes from arachidonic acid. Leukotrienes have an important role in the inflammatory process, and hence, inhibitors of 5-LOX exert an anti-inflammatory action [ 54 ]. By contrast, hydroxamic acid 7 was 10 times less potent than both celecoxib and hydroxamic acid 8 , with a COX-2 selectivity comparable to that of celecoxib. Both hydroxamic acid derivatives were effective anti-inflammatory agents in vivo , with ED 50 values of 23.1 μM and 24.5 μM for compounds 7 and 8 , respectively, compared to 10.8 μM for celecoxib and 128 μM for aspirin. It is worth mentioning that these compounds also exhibited a time-dependent release of nitric oxide, which leads to a GIT-safe profile.
- Biava et al. (2012) reported a novel class of diarylpyrrole acetic acid derivatives, which possess the typical scaffold of classic selective COX-2 inhibitors ( Figure 11 ). The researchers showed that 9 , its hydroxyethyl and hydroxypropyl ester intermediates ( 10a and 10b ), and their corresponding nitrate esters ( 11a and 11b ) were all potent selective COX-2 inhibitors in vitro (IC 50 = 0.019-0.083 μM) and considerably more selective than celecoxib. None of the esters ( 10a , 10b , 11a , and 11b ) were claimed by the authors to be prodrugs of 9 . Furthermore, molecular docking experiments have shown that the alcohol group in 10a and 10b and one of the oxygen atoms of the nitrates in 11a and 11b form one or more hydrogen bonds with amino acid side chains in the active site of COX-2. Furthermore, the nitrate ester 11a exhibited an in vitro vasorelaxing effect comparable to that of nitroglycerin [ 55 ]. This vasorelaxing effect can be considered the rationale behind designing NO-Coxibs.
- Finally, an example that previously has gone unnoticed is the furoxan-containing ester of aspirin 12 ( Figure 12 ), which possesses pronounced anti-inflammatory and anti-platelet action in vivo and very low gastric side effects without any detectable hydrolysis to aspirin [ 42 ].
2.3. The Role of the Linker in NO-NSAIDs
2.4. no-nsaids and in vivo hydrolysis, 3. anticholinergic nsaids and achei-nsaids, 3.1. introduction, 3.2. anticholinergic nsaids, 3.3. achei-nsaids, 4. phospho-nsaids, 4.1. introduction, 4.2. mechanism of action phospho-nsaids, 5. miscellaneous agents, 5.1. tempo-nsaids, 5.2. hs-nsaids, 6. conclusions.
- Albert, A. Chemical Aspects of Selective Toxicity. Nature 1958 , 182 , 421–423. [ Google Scholar ]
- Li, X. Oral Bioavailability: Basic Principles, Advanced Concepts, and Applications ; John Wiley & Sons: Hoboken, NJ, USA, 2011; p. 448. [ Google Scholar ]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov 2008 , 7 , 255–270. [ Google Scholar ]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev 2011 , 63 , 750–771. [ Google Scholar ]
- Testa, B. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol 2009 , 13 , 338–344. [ Google Scholar ]
- Das, N.; Dhanawat, M.; Dash, B.; Nagarwal, R.C.; Shrivastava, S.K. Codrug: An efficient approach for drug optimization. Eur. J. Pharm. Sci 2010 , 41 , 571–588. [ Google Scholar ]
- Qandil, A.; Al-Nabulsi, S.; Al-Taani, B.; Tashtoush, B. Synthesis of piperazinylalkyl ester prodrugs of ketorolac and their in vitro evaluation for transdermal delivery. Drug Dev. Ind. Pharm 2008 , 34 , 1054–1063. [ Google Scholar ]
- Abu Zanat, F.Z.; Qandil, A.M.; Tashtoush, B.M. A promising codrug of nicotinic acid and ibuprofen for managing dyslipidemia. I: Synthesis and in vitro evaluation. Drug Dev. Ind. Pharm 2011 , 37 , 1090–1099. [ Google Scholar ]
- Chung, M.C.; Bosquesi, P.L.; dos Santos, J.L. A Prodrug approach to improve the physico-chemical properties and decrease the genotoxicity of nitro compounds. Curr. Pharm. Des 2011 , 17 , 3515–3526. [ Google Scholar ]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons Learned from Marketed and Investigational Prodrugs. J. Med. Chem 2004 , 47 , 2393–2404. [ Google Scholar ]
- Stella, V.J. Prodrugs: Some thoughts and current issues. J. Pharm. Sci 2010 , 99 , 4755–4765. [ Google Scholar ]
- Qandil, A.M.; Rezigue, M.M.; Tashtoush, B.M. Synthesis, characterization and in vitro hydrolysis of a gemfibrozil-nicotinic acid codrug for improvement of lipid profile. Eur. J. Pharm. Sci 2011 , 43 , 99–108. [ Google Scholar ]
- Dhaneshwar, S.; Vadnerkar, G. Rational Design and Development of Colon-Specific Prodrugs. Curr. Top. Med. Chem 2011 , 11 , 2318–2345. [ Google Scholar ]
- Jana, S.; Mandlekar, S.; Marathe, P. Prodrug design to improve pharmacokinetic and drug delivery properties: Challenges to the discovery scientists. Curr. Med. Chem 2010 , 17 , 3874–3908. [ Google Scholar ]
- Teagarden, D.L.; Nema, S. Case Study: Parecoxib: A Prodrug of Valdecoxib Prodrugs ; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 1335–1346. [ Google Scholar ]
- Melnikova, I. Pain market. Nat. Rev. Drug Discov 2010 , 9 , 589–590. [ Google Scholar ]
- Laine, L. The gastrointestinal effects of nonselective NSAIDs and COX-2—selective inhibitors. Semin. Arthritis Rheumatism 2002 , 32 , 25–32. [ Google Scholar ]
- Gwaltney-Brant, S.M. Nonsteroidal Anti-Inflammatory Drug-Induced Toxicity. In Comprehensive Toxicology , 2nd ed; Charlene, A.M., Ed.; Elsevier: Oxford, UK, 2010; pp. 159–161. [ Google Scholar ]
- Lanas, A.; Garcia-Tell, G.; Armada, B.; Oteo-Alvaro, A. Prescription patterns and appropriateness of NSAID therapy according to gastrointestinal risk and cardiovascular history in patients with diagnoses of osteoarthritis. BMC Med 2011 , 9 , 38. [ Google Scholar ]
- Marnett, L.J. Mechanisms of cyclooxygenase-2 inhibition and cardiovascular side effects—The plot thickens. Cancer Prevention Res 2009 , 2 , 288–290. [ Google Scholar ]
- Bäck, M.; Yin, L.; Ingelsson, E. Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur. Heart J 2011 , 33 , 1928–1933. [ Google Scholar ]
- Duggan, D.E. Sulindac: Therapeutic Implications of the Prodrug/Pharmacophore Equilibrium. Drug Metab. Rev 1981 , 12 , 325–337. [ Google Scholar ]
- Sloan, K.B.; Wasdo, S. Designing for topical delivery: Prodrugs can make the difference. Med. Res. Rev 2003 , 23 , 763–793. [ Google Scholar ]
- Maag, H. Prodrugs of Carboxylic Acids. In Prodrugs ; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New York, NY, USA, 2007; pp. 703–729. [ Google Scholar ]
- Pavan, B.; Dalpiaz, A.; Ciliberti, N.; Biondi, C.; Manfredini, S.; Vertuani, S. Progress in drug delivery to the central nervous system by the prodrug approach. Molecules 2008 , 13 , 1035–1065. [ Google Scholar ]
- Halen, P.K.K.; Murumkar, P.R.; Giridhar, R.; Yadav, M.R. Prodrug designing of NSAIDs. Mini Rev. Med. Chem 2009 , 9 , 124–139. [ Google Scholar ]
- Müller, C.E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodiversity 2009 , 6 , 2071–2083. [ Google Scholar ]
- Koc, E.; Kucukguzel, S.G. Medicinal chemistry and anti-inflammatory activity of nitric oxide-releasing NSAI drugs. Mini Rev. Med. Chem 2009 , 9 , 611–619. [ Google Scholar ]
- Karim, A.; Laurent, A.; Slater, M.; Kuss, M.; Qian, J.; Crosby-Sessoms, S.; Hubbard, R. A Pharmacokinetic study of intramuscular (i.m) Parecoxib Sodium in normal subjects. Clin. Pharmacol 2001 , 41 , 1111–1119. [ Google Scholar ]
- Mamidi, R.; Mullangi, R.; Kota, J.; Bhamidipati, R.; Khan, A.; Katneni, K.; Datla, S.; Singh, S.; Rao, K.; Rao, C.; et al. Pharmacological and pharmacokinetic evaluation of celecoxib prodrugs in rats. Biopharm. Drug Dispos 2002 , 23 , 273–282. [ Google Scholar ]
- Qandil, A.M.; El Mohtadi, F.H.; Tashtoush, B.M. Chemical and in vitro enzymatic stability of newly synthesized celecoxib lipophilic and hydrophilic amides. Int. J. Pharm 2011 , 416 , 85–96. [ Google Scholar ]
- Lanas, A. Role of nitric oxide in the gastrointestinal tract. Arthritis Res. Ther 2008 , 10 , 1–6. [ Google Scholar ]
- Daff, S. NO synthase: Structures and mechanisms. Nitric Oxide 2010 , 23 , 1–11. [ Google Scholar ]
- Ruan, C.-H.; So, S.-P.; Ruan, K.-H. Inducible COX-2 dominates over COX-1 in prostacyclin biosynthesis: Mechanisms of COX-2 inhibitor risk to heart disease. Life Sci 2011 , 88 , 24–30. [ Google Scholar ]
- Sharma, S.K.; Al-Hourani, B.J.; Wuest, M.; Mane, J.Y.; Tuszynski, J.; Baracos, V.; Suresh, M.; Wuest, F. Synthesis and evaluation of fluorobenzoylated di- and tripeptides as inhibitors of cyclooxygenase-2 (COX-2). Bioorg. Med. Chem 2012 , 20 , 2221–2226. [ Google Scholar ]
- Ongini, E.; Bolla, M. Nitric-oxide based nonsteroidal anti-inflammatory agents. Drug Discovery Today 2006 , 3 , 395–400. [ Google Scholar ]
- Wong, P.S.-Y.; Fukuto, J.M. Reaction of organic nitrate esters ands-nitrosothiols with reduced flavins: A possible mechanism of bioactivation. Drug Metab. Dispos 1999 , 27 , 502–509. [ Google Scholar ]
- Minamiyama, Y.; Takemura, S.; Akiyama, T.; Imaoka, S.; Inoue, M.; Funae, Y.; Okada, S. Isoforms of cytochrome P450 on organic nitrate-derived nitric oxide release in human heart vessels. FEBS Lett 1999 , 452 , 165–169. [ Google Scholar ]
- Wallace, J.L.; Reuter, B.; Cicala, C.; McKnight, W.; Grisham, M.B.; Cirino, G. Novel nonsteroidal anti-inflammatory drug derivatives with markedly reduced ulcerogenic properties in the rat. Gastroenterology 1994 , 107 , 173–179. [ Google Scholar ]
- Bandarage, U.K.; Chen, L.; Fang, X.; Garvey, D.S.; Glavin, A.; Janero, D.R.; Letts, L.G.; Mercer, G.J.; Saha, J.K.; Schroeder, J.D.; et al. Nitrosothiol esters of diclofenac: Synthesis and pharmacological characterization as gastrointestinal-sparing prodrugs. J. Med. Chem 2000 , 43 , 4005–4016. [ Google Scholar ]
- Gilmer, J.F.; Moriarty, L.M.; McCafferty, D.F.; Clancy, J.M. Synthesis, hydrolysis kinetics and anti-platelet effects of isosorbide mononitrate derivatives of aspirin. Eur. J. Pharm. Sci 2001 , 14 , 221–227. [ Google Scholar ]
- Cena, C.; Lolli, M.L.; Lazzarato, L.; Guaita, E.; Morini, G.; Coruzzi, G.; McElroy, S.P.; Megson, I.L.; Fruttero, R.; Gasco, A. Antiinflammatory, gastrosparing, and antiplatelet properties of new no-donor esters of aspirin. J. Med. Chem 2003 , 46 , 747–754. [ Google Scholar ]
- Velázquez, C.; Rao, P.N.P.; Knaus, E.E. Novel nonsteroidal antiinflammatory drugs possessing a nitric oxide donor diazen-1-ium-1,2-diolate moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem 2005 , 48 , 4061–4067. [ Google Scholar ]
- Ranatunge, R.R.; Augustyniak, M.E.; Dhawan, V.; Ellis, J.L.; Garvey, D.S.; Janero, D.R.; Letts, L.G.; Richardson, S.K.; Shumway, M.J.; Trocha, A.M.; et al. Synthesis and anti-inflammatory activity of a series of N -substituted naproxen glycolamides: Nitric oxide-donor naproxen prodrugs. Bioorg. Med. Chem 2006 , 14 , 2589–2599. [ Google Scholar ]
- Velázquez, C.; Chen, Q.-H.; Citro, M.L.; Keefer, L.K.; Knaus, E.E. Second-Generation aspirin and indomethacin prodrugs possessing an O 2-(acetoxymethyl)-1-(2-carboxypyrrolidin-1-yl)diazenium-1,2-diolate nitric oxide donor moiety: Design, synthesis, biological evaluation, and nitric oxide release studies. J. Med. Chem 2008 , 51 , 1954–1961. [ Google Scholar ]
- De Carvalho, P.S.; Maróstica, M.; Gambero, A.; Pedrazzoli, J., Jr. Synthesis and pharmacological characterization of a novel nitric oxide-releasing diclofenac derivative containing a benzofuroxan moiety. Eur. J. Med. Chem. 2010 , 45 , 2489–2493. [ Google Scholar ]
- Huang, Z.; Velázquez, C.; Abdellatif, K.; Chowdhury, M.A.; Reisz, J.; DuMond, J.; King, S.B.; Knaus, E.E. Ethanesulfohydroxamic acid ester prodrugs of nonsteroidal anti-inflammatory drugs (nsaids): Synthesis, nitric oxide and nitroxyl release, cyclooxygenase inhibition, anti-inflammatory, and ulcerogenicity index studies. J. Med. Chem 2011 , 54 , 1356–1364. [ Google Scholar ]
- Chattopadhyay, M.; Velazquez, C.A.; Pruski, A.; Nia, K.V.; Abdellatif, K.R.; Keefer, L.K.; Kashfi, K. Comparison between 3-nitrooxyphenyl acetylsalicylate (NO-ASA) and O 2-(acetylsalicyloxymethyl)-1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate (NONO-ASA) as safe anti-Inflammatory, analgesic, antipyretic, antioxidant prodrugs. J. Pharmacol. Exp. Ther 2010 , 335 , 443–450. [ Google Scholar ]
- Jain, S.; Tran, S.; El Gendy, M.A.M.; Kashfi, K.; Jurasz, P.; Velázquez-Martínez, C.A. Nitric oxide release is not required to decrease the ulcerogenic profile of nonsteroidal anti-inflammatory drugs. J. Med. Chem 2011 , 55 , 688–696. [ Google Scholar ]
- Maragos, C.M.; Morley, D.; Wink, D.A.; Dunams, T.M.; Saavedra, J.E.; Hoffman, A.; Bove, A.A.; Isaac, L.; Hrabie, J.A.; Keefer, L.K. Complexes of .NO with nucleophiles as agents for the controlled biological release of nitric oxide. Vasorelaxant effects. J. Med. Chem 1991 , 34 , 3242–3247. [ Google Scholar ]
- Pathan, A.; Karwa, M.; Pamidiboina, V.; Deshattiwar, J.; Deshmukh, N.; Gaikwad, P.; Mali, S.; Desai, D.; Dhiman, M.; Mariappan, T.T.; et al. Oral bioavailability, efficacy and gastric tolerability of P2026, a novel nitric oxide-releasing diclofenac in rat. Inflammopharmacology 2010 , 18 , 157–168. [ Google Scholar ]
- Abdellatif, K.R.; Chowdhury, M.A.; Dong, Y.; Das, D.; Yu, G.; Velazquez, C.A.; Suresh, M.R.; Knaus, E.E. Dinitroglyceryl and diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of aspirin, indomethacin and ibuprofen: synthesis, biological evaluation and nitric oxide release studies. Bioorg. Med. Chem. Lett 2009 , 19 , 3014–3018. [ Google Scholar ]
- Kaur, J.; Bhardwaj, A.; Huang, Z.; Knaus, E.E. Aspirin analogues as dual cyclooxygenase-2/5- lipoxygenase inhibitors: Synthesis, nitric oxide release, molecular modeling, and biological evaluation as anti-inflammatory agents. ChemMedChem 2012 , 7 , 144–150. [ Google Scholar ]
- Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.-P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheumatic Dis 2003 , 62 , 501–509. [ Google Scholar ]
- Biava, M.; Porretta, G.C.; Poce, G.; Battilocchio, C.; Alfonso, S.; Rovini, M.; Valenti, S.; Giorgi, G.; Calderone, V.; Martelli, A.; et al. Novel analgesic/anti-inflammatory agents: Diarylpyrrole acetic esters endowed with nitric oxide releasing properties. J. Med. Chem 2011 , 54 , 7759–7771. [ Google Scholar ]
- Fabbri, F.; Brigliadori, G.; Ulivi, P.; Tesei, A.; Vannini, I.; Rosetti, M.; Bravaccini, S.; Amadori, D.; Bolla, M.; Zoli, W. Pro-apoptotic effect of a nitric oxide-donating NSAID, NCX 4040, on bladder carcinoma cells. Apoptosis 2005 , 10 , 1095–1103. [ Google Scholar ]
- Tesei, A.; Rosetti, M.; Ulivi, P.; Fabbri, F.; Medri, L.; Vannini, I.; Bolla, M.; Amadori, D.; Zoli, W. Study of molecular mechanisms of pro-apoptotic activity of NCX 4040, a novel nitric oxide-releasing aspirin, in colon cancer cell lines. J. Transl. Med 2007 , 5 , 52. [ Google Scholar ]
- Tesei, A.; Ulivi, P.; Fabbri, F.; Rosetti, M.; Leonetti, C.; Scarsella, M.; Zupi, G.; Amadori, D.; Bolla, M.; Zoli, W. In vitro and in vivo evaluation of NCX 4040 cytotoxic activity in human colon cancer cell lines. J. Transl. Med 2005 , 3 , 7. [ Google Scholar ]
- Tesei, A.; Zoli, W.; Fabbri, F.; Leonetti, C.; Rosetti, M.; Bolla, M.; Amadori, D.; Silvestrini, R. NCX 4040, an NO-donating acetylsalicylic acid derivative: Efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008 , 19 , 225–236. [ Google Scholar ]
- Rosetti, M.; Tesei, A.; Ulivi, P.; Fabbri, F.; Vannini, I.; Brigliadori, G.; Amadori, D.; Bolla, M.; Zoli, W. Molecular characterization of cytotoxic and resistance mechanisms induced by NCX 4040, a novel NO-NSAID, in pancreatic cancer cell lines. Apoptosis 2006 , 11 , 1321–1330. [ Google Scholar ]
- Hulsman, N.; Medema, J.P.; Bos, C.; Jongejan, A.; Leurs, R.; Smit, M.J.; de Esch, I.J.P.; Richel, D.; Wijtmans, M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem 2007 , 50 , 2424–2431. [ Google Scholar ]
- Kashfi, K.; Rigas, B. The mechanism of action of nitric oxide-donating aspirin. Biochem. Biophys. Res. Commun 2007 , 358 , 1096–1101. [ Google Scholar ]
- Nemmani, K.V.S.; Mali, S.V.; Borhade, N.; Pathan, A.R.; Karwa, M.; Pamidiboina, V.; Senthilkumar, S.P.; Gund, M.; Jain, A.K.; Mangu, N.K.; et al. NO-NSAIDs: Gastric-sparing nitric oxide-releasable prodrugs of non-steroidal anti-inflammatory drugs. Bioorg. Med. Chem. Lett 2009 , 19 , 5297–5301. [ Google Scholar ]
- Lazzarato, L.; Chegaev, K.; Marini, E.; Rolando, B.; Borretto, E.; Guglielmo, S.; Joseph, S.; di Stilo, A.; Fruttero, R.; Gasco, A. New nitric oxide or hydrogen sulfide releasing aspirins. J. Med. Chem 2011 , 54 , 5478–5484. [ Google Scholar ]
- Wey, S.-J.; Augustyniak, M.E.; Cochran, E.D.; Ellis, J.L.; Fang, X.; Garvey, D.S.; Janero, D.R.; Letts, L.G.; Martino, A.M.; Melim, T.L.; et al. Structure-Based design, synthesis, and biological evaluation of indomethacin derivatives as cyclooxygenase-2 inhibiting nitric oxide donors. J. Med. Chem 2007 , 50 , 6367–6382. [ Google Scholar ]
- Downing, J.E.G.; Madden, J.C.; Ingram, M.J.; Rostron, C. Gastric and thymic assay of acute oral treatment of rats with nitric oxide esters of ibuprofen or indomethacin. Biochem. Biophys. Res. Commun 2005 , 334 , 646–653. [ Google Scholar ]
- Yang, C.-F.; Zhang, Y.-Y.; Yang, B.; Li, P.-F.; Zhuang, D.-Y. Synthesis of indomethacin derivatives and their anti-inflammatory activities. Chin. New Drugs J 2004 , 9 , 818–2106. [ Google Scholar ]
- Schiefer, I.T.; Abdul-Hay, S.; Wang, H.; Vanni, M.; Qin, Z.; Thatcher, G.R.J. Inhibition of amyloidogenesis by nonsteroidal anti-inflammatory drugs and their hybrid nitrates. J. Med. Chem 2011 , 54 , 2293–2306. [ Google Scholar ]
- Schubert, M.L.; Peura, D.A. Control of gastric acid secretion in health and disease. Gastroenterology 2008 , 134 , 1842–1860. [ Google Scholar ]
- Ehlert, F.J.; Pak, K.J.; Griffin, M.T. Muscarinic Agonists and Antagonists: Effects on Gastrointestinal Function Muscarinic Receptors ; Fryer, A.D., Christopoulos, A., Nathanson, N.M., Eds.; Springer: Berlin, Germany, 2012; Volume 208, pp. 343–374. [ Google Scholar ]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Synthesis and pharmacological evaluation of some dual-acting amino-alcohol ester derivatives of flurbiprofen and 2-[1,1′-biphenyl-4-yl]acetic acid: A potential approach to reduce local gastrointestinal toxicity. Chem. Biodiversity 2006 , 3 , 1238–1248. [ Google Scholar ]
- Sato, N.; Kawano, S.; Tsuji, S.; Ogihara, T.; Yamada, S. Gastric blood flow in ulcer diseases. Scand. J. Gastroenterol 1995 , 30 , 14–20. [ Google Scholar ]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Substituted aminoalcohol ester analogs of indomethacin with reduced toxic effects. Med. Chem. Res 2007 , 16 , 101–111. [ Google Scholar ]
- Halen, P.K.K.; Chagti, K.K.; Giridhar, R.; Yadav, M.R. Combining anticholinergic and anti-inflammatory activities into a single moiety: A novel approach to reduce gastrointestinal toxicity of ibuprofen and ketoprofen. Chem. Biol. Drug Des 2007 , 70 , 450–455. [ Google Scholar ]
- Cowan, F.M.; Broomfield, C.A.; Lenz, D.E.; Smith, W.J. Putative role of proteolysis and inflammatory response in the toxicity of nerve and blister chemical warfare agents: Implications for multi-threat medical countermeasures. J. Appl. Toxicol 2003 , 23 , 177–186. [ Google Scholar ]
- Pita, R.; Vidal-Asensi, S. Cutaneous and systemic toxicology of vesicant (blister) warfare agents. Actas Dermo-Sifiliográficas (English Edition) 2010 , 101 , 7–18. [ Google Scholar ]
- Tracey, K.J. The inflammatory reflex. Nature 2002 , 420 , 853–859. [ Google Scholar ]
- Green, R.D.; Fleming, W.W. Agonist-Antagonist interactions in the normal and supersensitive nictitating membrane of the spinal cat. J. Pharmacol. Exp. Ther 1967 , 156 , 207–214. [ Google Scholar ]
- Kalgutkar, A.S.; Marnett, A.B.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J. Med. Chem 2000 , 43 , 2860–2870. [ Google Scholar ]
- Rautio, J.; Nevalainen, T.; Taipale, H.; Vepsäläinen, J.; Gynther, J.; Laine, K.; Järvinen, T. Piperazinylalkyl prodrugs of naproxen improve in vitro skin permeation. Eur. J. Pharm. Sci 2000 , 11 , 157–163. [ Google Scholar ]
- Young, S.; Fabio, K.; Guillon, C.; Mohanta, P.; Halton, T.A.; Heck, D.E.; Flowers, R.A., II; Laskin, J.D.; Heindel, N.D. Peripheral site acetylcholinesterase inhibitors targeting both inflammation and cholinergic dysfunction. Bioorg. Med. Chem. Lett 2010 , 20 , 2987–2990. [ Google Scholar ]
- Young, S.; Fabio, K.; Huang, M.-T.; Saxena, J.; Harman, M.; Guillon, C.; Vetrano, A.; Heck, D.; Flowers, R.; Heindel, N.; et al. Investigation of anticholinergic and non-steroidal anti-inflammatory prodrugs which reduce chemically induced skin inflammation. J. Appl. Toxicol 2012 , 32 , 135–141. [ Google Scholar ]
- Huang, L.; Mackenzie, G.; Sun, Y.; Ouyang, N.; Xie, G.; Vrankova, K.; Komninou, D.; Rigas, B. Chemotherapeutic properties of phospho-nonsteroidal anti-inflammatory drugs, a new class of anticancer compounds. Cancer Res 2011 , 71 , 7617–7627. [ Google Scholar ]
- Huang, L.; Mackenzie, G.; Ouyang, N.; Sun, Y.; Xie, G.; Johnson, F.; Komninou, D.; Rigas, B. The novel phospho-non-steroidal anti-inflammatory drugs, OXT-328, MDC-22 and MDC-917, inhibit adjuvant-induced arthritis in rats. Br. J. Pharmacol 2011 , 162 , 1521–1533. [ Google Scholar ]
- Sun, Y.; Huang, L.; Mackenzie, G.G.; Rigas, B. Oxidative stress mediates through apoptosis the anticancer effect of phospho-nonsteroidal anti-inflammatory drugs: Implications for the role of oxidative stress in the action of anticancer agents. J. Pharmacol. Exp.Ther 2011 , 338 , 775–783. [ Google Scholar ]
- Huang, L.; Zhu, C.; Sun, Y.; Xie, G.; Mackenzie, G.; Qiao, G.; Komninou, D.; Rigas, B. Phospho-sulindac (OXT-922) inhibits the growth of human colon cancer cell lines: A redox/polyamine dependent effect. Carcinogenesis 2010 , 31 , 1982–1990. [ Google Scholar ]
- Xie, G.; Sun, Y.; Nie, T.; Mackenzie, G.G.; Huang, L.; Kopelovich, L.; Komninou, D.; Rigas, B. Phospho-Ibuprofen (MDC-917) is a novel agent against colon cancer: efficacy, metabolism, and pharmacokinetics in mouse models. J. Pharmacol. Exp. Ther 2011 , 337 , 876–886. [ Google Scholar ]
- Wong, C.C.; Cheng, K.-W.; Xie, G.; Zhou, D.; Zhu, C.-H.; Constantinides, P.P.; Rigas, B. Carboxylesterases 1 and 2 hydrolyze phospho-nonsteroidal anti-inflammatory drugs: Relevance to their pharmacological activity. J. Pharmacol. Exp. Ther 2012 , 340 , 422–432. [ Google Scholar ]
- Zhao, W.; Mackenzie, G.G.; Murray, O.T.; Zhang, Z.; Rigas, B. Phosphoaspirin (MDC-43), a novel benzyl ester of aspirin, inhibits the growth of human cancer cell lines more potently than aspirin: A redox-dependent effect. Carcinogenesis 2009 , 30 , 512–519. [ Google Scholar ]
- Dufrasne, F.; Gelbcke, M.; Neve, J.; Kiss, R.; Kraus, J.-L. Quinone methides and their prodrugs: A subtle equilibrium between cancer promotion, prevention, and cure. Curr. Med. Chem 2011 , 18 , 3995–4011. [ Google Scholar ]
- Deguchi, H.; Yasukawa, K.; Yamasaki, T.; Mito, F.; Kinoshita, Y.; Naganuma, T.; Sato, S.; Yamato, M.; Ichikawa, K.; Sakai, K.; et al. Nitroxides prevent exacerbation of indomethacin-induced gastric damage in adjuvant arthritis rats. Free Radical Biol. Med 2011 , 51 , 1799–1805. [ Google Scholar ]
- Flores-Santana, W.; Moody, T.; Chen, W.; Gorczynski, M.J.; Shoman, M.E.; Velázquez, C.; Thetford, A.; Mitchell, J.B.; Cherukuri, M.K.; King, S.B.; et al. Nitroxide derivatives of non-steroidal anti-inflammatory drugs exert anti-inflammatory and superoxide dismutase scavenging properties in A459 cells. Br. J. Pharmacol 2012 , 165 , 1058–1067. [ Google Scholar ]
- Yokomizo, T.; Izumi, T.; Shimizu, T. Co-expression of two LTB4 receptors in human mononuclear cells. Life Sci 2001 , 68 , 2207–2212. [ Google Scholar ]
- Caliendo, G.; Cirino, G.; Santagada, V.; Wallace, J.L. Synthesis and biological effects of hydrogen sulfide (h 2 s): Development of h 2 s-releasing drugs as pharmaceuticals. J. Med. Chem 2010 , 53 , 6275–6286. [ Google Scholar ]
- Fiorucci, S.; Distrutti, E.; Cirino, G.; Wallace, J.L. The emerging roles of hydrogen sulfide in the gastrointestinal tract and liver. Gastroenterology 2006 , 131 , 259–271. [ Google Scholar ]
- Wallace, J.L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci 2007 , 28 , 501–505. [ Google Scholar ]
- Zanatta, S.D.; Jarrott, B.; Williams, S.J. Synthesis and preliminary pharmacological evaluation of aryl dithiolethiones with cyclooxygenase-2-selective inhibitory activity and hydrogen sulfide-releasing properties. Aust. J. Chem 2010 , 63 , 946–957. [ Google Scholar ]
- Kodela, R.; Chattopadhyay, M.; Kashfi, K. NOSH-Aspirin: A novel nitric oxide–hydrogen sulfide-releasing hybrid: A new class of anti-inflammatory pharmaceuticals. ACS Med. Chem. Lett 2012 , 3 , 257–262. [ Google Scholar ]
- Chattopadhyay, M.; Kodela, R.; Nath, N.; Dastagirzada, Y.M.; Velázquez-Martínez, C.A.; Boring, D.; Kashfi, K. Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: A general property and evidence of a tissue type-independent effect. Biochem. Pharmacol 2012 , 83 , 715–722. [ Google Scholar ]
- Chattopadhyay, M.; Kodela, R.; Olson, K.R.; Kashfi, K. NOSH–aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model. Biochem. Biophy. Res. Commun 2012 , 419 , 523–528. [ Google Scholar ]
Click here to enlarge figure
| Name | NO donor moiety | Equivalents of NO released | Examples |
|---|---|---|---|
| Nitrates | –ONO | 1 | |
| NONOate | 2 | ||
| Furoxan and Benzofuroxan | 1 | ||
| Sulfohydroxamic Acid | 1 | ||
| Nitrosothiol Esters | 1 |
| Compound | t , pH 7.4 (h) | % NSAID released in plasma after 2 h | pA | % inhibition of Edema Volume | UI |
|---|---|---|---|---|---|
| Ketoprofen | 87 | 0.882 | |||
| 21a–e | 16–64 | 58.9–79.6 | 5.28–6.43 | 70–89 | 0.306–0.376 |
| 21f | ND | ND | 7.58 | 86 | 0.299 |
| Flurbiprofen | 0 | 0.800 | |||
| 22a–e | 121–505 | 17.9–51.1 | 4.73–5.79 | 79–89 | 0.130–0.230 |
| 22f | ND | ND | 6.31 | 0 | 0.00 |
| Indomethacin | 45 | 0.55 | |||
| 23a–e | 34–99 | 49.6–79.9 | 4.49–5.19 | 17–46 | 0.130–0.320 |
| Atropine , , | 8.02 |
| Comd | % reduction in edema (Mouse ear vesicant model) [ ] | Acetylcholinesterase Inhibition [ ] | Plasma t (min) | Calculated LogP | |
|---|---|---|---|---|---|
| CEES | TPA | IC (μM) | |||
| 24a | 20 | 41 | 1.93 ± 0.64 | 204 | 7.37 |
| 25b | 45 | 70 | 0.83 ± 0.15 | 253 | 6.67 |
| 26a | 91 | 21 | 2.29 ± 0.94 | 468 | 7.87 |
| 27a | 90 | 24 | 0.51 ± 0.02 | 357 | 8.41 |
| 31a | 113 | 29 | 2.69 ± 0.15 | 111 | 7.52 |
| Tacrine | - | - | 0.055 ± 0.005 | - | - |
| DIC | 17 | 58 | - | - | - |
| IND | 46 | 55 | - | - | - |
| IBU | -15 | -33 | - | - | - |
| NAP | NS | * | 104 | - | - |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qandil, A.M. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. Int. J. Mol. Sci. 2012 , 13 , 17244-17274. https://doi.org/10.3390/ijms131217244
Qandil AM. Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review. International Journal of Molecular Sciences . 2012; 13(12):17244-17274. https://doi.org/10.3390/ijms131217244
Qandil, Amjad M. 2012. "Prodrugs of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs), More Than Meets the Eye: A Critical Review" International Journal of Molecular Sciences 13, no. 12: 17244-17274. https://doi.org/10.3390/ijms131217244
Article Metrics
Article access statistics, further information, mdpi initiatives, follow mdpi.
Subscribe to receive issue release notifications and newsletters from MDPI journals
Prodrugs: Solubility, Absorption, and Biostability Research Paper
- To find inspiration for your paper and overcome writer’s block
- As a source of information (ensure proper referencing)
- As a template for you assignment
The Solubility of Prodrugs
The absorption of prodrugs, biostability of prodrugs.
An effective prodrug needs to be soluble so that it can reach the target site and affect the desirable pharmacologic effects. Prodrugs are designed to circumvent drug limitations such as poor solubility in the aqueous and lipid phases. Therefore, most prodrugs are soluble in the body compartment where they are expected to affect their pharmacologic actions. The solubility of prodrugs can be altered by the addition of certain functional groups. For example, prodrug esters are designed to augment their lipophilic properties so that water-soluble drugs can easily traverse the cell membranes (Zawilska, Wojcieszak and Olejniczak 2). Their solubility in the aqueous phases is also improved by using phosphate esters and amides. In some cases, hydrophilic hydroxyl groups contain pharmacologically active substances. Acylation with aliphatic or aromatic carboxylic acids is carried out to improve their solubility in the lipid phase. To improve the water solubility of prodrugs, the hydroxyl groups of the parent drug are usually esterified with carboxylic acid products with other functional groups, for instance, amino or hydroxyl groups. These groups confer hydrophilicity or increased aqueous solubility to the prodrug molecules.
A novel approach to improving the solubility of prodrugs has been demonstrated in the hydrogenation of naproxen (an analgesic) to produce a hydrogelator, which improves its solubility and eliminates the need for a carrier molecule (Majumder et al. 10254).
The efficacy of prodrugs is determined by their ability to be absorbed through oral and non-oral routes. The rapid absorption of the drug into the target cells is often necessary for a faster pharmacotherapeutic action of a drug (Okudaira et al. 580). Therefore, the absorption of most prodrugs is modified to match the anticipated action of the active drug. The advancement of oral bioavailability of drugs is among the most notable innovations since the discovery of prodrugs, which is attributed to improving their oral absorption (Zawilska, Wojcieszak and Olejniczak 3). Cancer treatment has also recorded immense progress by improving the absorption of anticancer agents. For example, Capecitabine (5′-deoxy-5-fluorocytidine carbamate), which is a tripartite prodrug undergoes fast and extensive oral absorption following oral administration. Liver carboxylesterases then act on the drug to yield 5′-deoxy-5-fluorocytidine that is further deaminated in the liver and cancerous cells by cytidine deaminase (Zawilska, Wojcieszak and Olejniczak 8). Ultimately, thymidine phosphorylase converts the metabolite to an extremely cytotoxic 5’-fluorouracil within the tumors.
The active drug is liberated from its dormant form before, in the course of, or following the absorption of the prodrug. Certain drugs can only be freed after they have arrived at the targets of their actions (Zawilska, Wojcieszak, and Olejniczak 2). Additionally, a prodrug needs to amplify the bioavailability and therapeutic value of a parent drug. Therefore, the preferred biostability of a prodrug varies with the site of release of the active drug. For prodrugs that are liberated after reaching their target sites, they should remain stable to ensure maximum absorption into the target sites. The biostability of prodrugs also aims at reducing presystemic metabolism, to enhance time rundown, elevating organ or tissue-selective supply of the active compound.
The biostability of prodrugs is influenced by the ease with which enzymes act on the prodrug to liberate the active compound. For example, the esterification of prodrugs with phosphate groups increases their biostability because endogenous phosphatases act rapidly on phosphate prodrugs. The functional groups present on the prodrug also determine the biostability of the drug. For example, drugs that require stability in the aqueous phase are formulated as phosphate prodrugs because amino acid ester exhibit inferior aqueous stability while amide prodrugs undergo partial in vivo bioconversion (Huttunen, Raunio, and Rautio 755).
Huttunen, Kristiina M., Hannu Raunio, and Jarkko Rautio. “Prodrugs—from Serendipity to Rational Design.” Pharmacological Reviews 63.3(2011):750–771. Print.
Majumder, Joydeb Mahua Rani Das, Jolly Deb, Siddhartha Sankar Jana, and Parthasarathi Dastidar. “β‑Amino Acid and Amino-Alcohol Conjugation of a Nonsteroidal Anti-Inflammatory Drug (NSAID) Imparts Hydrogelation Displaying Remarkable Biostability, Biocompatibility, and Anti-Inflammatory Properties.” Langmuir 29.32(2013): 10254−10263. Print.
Okudaira, Noriko, Tomoko Tatebayashi, Graham C. Speirs, Izumi Komiya, and Yuichi Sugiyama. “A Study of the Intestinal Absorption of an Ester-Type Prodrug, ME3229, in Rats: Active Efflux Transport as a Cause of Poor Bioavailability of the Active Drug.” The Journal of Pharmacology and Experimental Therapeutics 294.2(2000):580-587. Print.
Zawilska, Jolanta B., Jakub Wojcieszak, and Agnieszka B. Olejniczak. “Prodrugs: A Challenge for the Drug Development.” Pharmacological Reports 65.1(2013):1–14. Print.
- Compounding Pharmacy Industry
- Neurotransmitter Transporters-Based Psychiatric Treatment
- Solubility of Potassium Nitrate
- Nitroglycerine's Routes of Administration
- KHT Molar Solubility Experiment
- Separation of the Constituents of an Analgesic Preparation
- Antiviral Drugs. Healthcare
- Food Security: Limiting the Use of Antibiotics to Reduce or Slow the Antibiotic Resistance
- Legal and Ethical Considerations: PharmCARE
- The Pros and Cons of OTC Acid-Reducing Agents
- Chicago (A-D)
- Chicago (N-B)
IvyPanda. (2022, April 18). Prodrugs: Solubility, Absorption, and Biostability. https://ivypanda.com/essays/prodrugs-solubility-absorption-and-biostability/
"Prodrugs: Solubility, Absorption, and Biostability." IvyPanda , 18 Apr. 2022, ivypanda.com/essays/prodrugs-solubility-absorption-and-biostability/.
IvyPanda . (2022) 'Prodrugs: Solubility, Absorption, and Biostability'. 18 April.
IvyPanda . 2022. "Prodrugs: Solubility, Absorption, and Biostability." April 18, 2022. https://ivypanda.com/essays/prodrugs-solubility-absorption-and-biostability/.
1. IvyPanda . "Prodrugs: Solubility, Absorption, and Biostability." April 18, 2022. https://ivypanda.com/essays/prodrugs-solubility-absorption-and-biostability/.
Bibliography
IvyPanda . "Prodrugs: Solubility, Absorption, and Biostability." April 18, 2022. https://ivypanda.com/essays/prodrugs-solubility-absorption-and-biostability/.
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 02 April 2024
The landscape of small-molecule prodrugs
- Zachary Fralish ORCID: orcid.org/0000-0001-6293-1730 1 ,
- Ashley Chen 2 ,
- Shaharyar Khan 3 ,
- Pei Zhou ORCID: orcid.org/0000-0002-7823-3416 4 &
- Daniel Reker ORCID: orcid.org/0000-0003-4789-7380 1
Nature Reviews Drug Discovery volume 23 , pages 365–380 ( 2024 ) Cite this article
10k Accesses
3 Citations
84 Altmetric
Metrics details
- Data mining
- Drug delivery
- Drug safety
- Pharmacokinetics
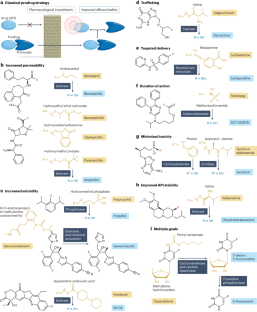
Prodrugs are derivatives with superior properties compared with the parent active pharmaceutical ingredient (API), which undergo biotransformation after administration to generate the API in situ. Although sharing this general characteristic, prodrugs encompass a wide range of different chemical structures, therapeutic indications and properties. Here we provide the first holistic analysis of the current landscape of approved prodrugs using cheminformatics and data science approaches to reveal trends in prodrug development. We highlight rationales that underlie prodrug design, their indications, mechanisms of API release, the chemistry of promoieties added to APIs to form prodrugs and the market impact of prodrugs. On the basis of this analysis, we discuss strengths and limitations of current prodrug approaches and suggest areas for future development.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
24,99 € / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

The evolution of commercial drug delivery technologies

Molecular chameleons in drug discovery

Foundations of gastrointestinal-based drug delivery and future developments
Rautio, J., Meanwell, N. A., Di, L. & Hageman, M. J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 17 , 559–587 (2018).
Article CAS PubMed Google Scholar
Mitchell, M. J. et al. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 20 , 101–124 (2021).
Narang, A. S. & Boddu, S. H. S. in Excipient Applications in Formulation Design and Drug Delivery (eds Narang, A. S. & Boddu, S. H. S) 1–10 (Springer, 2015).
Zhao, Z., Ukidve, A., Kim, J. & Mitragotri, S. Targeting strategies for tissue-specific drug delivery. Cell 181 , 151–167 (2020).
Elsharkasy, O. M. et al. Extracellular vesicles as drug delivery systems: why and how? Adv. Drug Deliv. Rev. 159 , 332–343 (2020).
Huttunen, K. M., Raunio, H. & Rautio, J. Prodrugs—from serendipity to rational design. Pharmacol. Rev. 63 , 750–771 (2011).
Najjar, A. & Karaman, R. The prodrug approach in the era of drug design. Expert Opin. Drug. Deliv. 16 , 1–5 (2018).
Article PubMed Google Scholar
Jordheim, L. P., Durantel, D., Zoulim, F. & Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 12 , 447–464 (2013).
Wang, Y. et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 48 , D1031–D1041 (2020).
CAS PubMed Google Scholar
Degtyarenko, K. et al. ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 36 , D344–D350 (2007).
Article PubMed PubMed Central Google Scholar
Mullard, A. 2022 FDA approvals. Nat. Rev. Drug Discov. 22 , 83–88 (2023).
Mullard, A. 2021 FDA approvals. Nat. Rev. Drug Discov. 21 , 83–98 (2022).
Mullard, A. 2020 FDA drug approvals. Nat. Rev. Drug Discov. 20 , 85–91 (2021).
Mullard, A. 2019 FDA drug approvals. Nat. Rev. Drug Discov. 19 , 79–84 (2020).
Wishart, D. S. et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 46 , D1074–D1082 (2018).
Zhou, Y. et al. Therapeutic target database update 2022: facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 50 , D1398–D1407 (2022).
Sugawara, M. et al. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J. Pharm. Sci. 89 , 781–789 (2000).
Peppercorn, M. A. Sulfasalazine: pharmacology, clinical use, toxicity, and related new drug development. Ann. Intern. Med. 101 , 377–386 (1984).
Asaki, T. et al. Selexipag: an oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J. Med. Chem. 58 , 7128–7137 (2015).
Lee, W. A. & Cheng, A. K. Tenofovir alafenamide fumarate. Antivir. Ther. 27 , 13596535211067600 (2022).
Jankovic, J. Dopamine depleters in the treatment of hyperkinetic movement disorders. Expert Opin. Pharmacother. 17 , 2461–2470 (2016).
Reigner, B., Blesch, K. & Weidekamm, E. Clinical pharmacokinetics of capecitabine. Clin. Pharmacokinet. 40 , 85–104 (2001).
Aljuffali, I. A., Lin, C.-F., Chen, C.-H. & Fang, J.-Y. The codrug approach for facilitating drug delivery and bioactivity. Expert Opin. Drug Deliv. 13 , 1311–1325 (2016).
Friedel, H. A., Campoli-Richards, D. M. & Goa, K. L. Sultamicillin. Drugs 37 , 491–522 (1989).
Mueller, C. E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodivers. 6 , 2071–2083 (2009).
Article CAS Google Scholar
Barlow, N., Chalmers, D. K., Williams-Noonan, B. J., Thompson, P. E. & Norton, R. S. Improving membrane permeation in the beyond rule-of-five space by using prodrugs to mask hydrogen bond donors. ACS Chem. Biol. 15 , 2070–2078 (2020).
Murakami, T. A minireview: usefulness of transporter-targeted prodrugs in enhancing membrane permeability. J. Pharm. Sci. 105 , 2515–2526 (2016).
Camp, D., Garavelas, A. & Campitelli, M. Analysis of physicochemical properties for drugs of natural origin. J. Nat. Prod. 78 , 1370–1382 (2015).
Hall, B. S. & Wilkinson, S. R. Activation of benznidazole by trypanosomal type I nitroreductases results in glyoxal formation. Antimicrob. Agents Chemother. 56 , 115–123 (2012).
Article CAS PubMed PubMed Central Google Scholar
Jubeh, B., Breijyeh, Z. & Karaman, R. Antibacterial prodrugs to overcome bacterial resistance. Molecules 25 , 1543 (2020).
Lin, D. et al. Bacterial-based cancer therapy: an emerging toolbox for targeted drug/gene delivery. Biomaterials 277 , 121124 (2021).
Li, Y., Zhao, L. & Li, X.-F. Targeting hypoxia: hypoxia-activated prodrugs in cancer therapy. Front. Oncol. 11 , 700407 (2021).
Peiró Cadahía, J., Previtali, V., Troelsen, N. S. & Clausen, M. H. Prodrug strategies for targeted therapy triggered by reactive oxygen species. Medchemcomm 10 , 1531–1549 (2019).
Padilla, A. M. et al. Discovery of an orally active benzoxaborole prodrug effective in the treatment of Chagas disease in non-human primates. Nat. Microbiol. 7 , 1536–1546 (2022).
Cundy, K. C. et al. XP13512 [(±)-1-([(α-Isobutanoyloxyethoxy) carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther. 311 , 315–323 (2004).
Salem, A. H. et al. Expanding the repertoire for “Large small molecules”: prodrug abbv-167 efficiently converts to venetoclax with reduced food effect in healthy volunteers. Mol. Cancer Ther. 20 , 999–1008 (2021).
Halpern, L. The transfer of inorganic phosphorus across the red blood cell membrane. J. Biol. Chem. 114 , 747–770 (1936).
Wiemer, A. J. Metabolic efficacy of phosphate prodrugs and the remdesivir paradigm. ACS Pharmacol. Transl. Sci. 3 , 613–626 (2020).
Ohwada, J. et al. Design, synthesis and antifungal activity of a novel water soluble prodrug of antifungal triazole. Bioorg. Med. Chem. Lett. 13 , 191–196 (2003).
Sawada, S. et al. Synthesis and antitumor activity of 20 (S)-camptothecin derivatives: Carbamate-linked, water-soluble derivaties of 7-ethyl-10-hydroxycamptothecin. Chem. Pharm. Bull. 39 , 1446–1454 (1991).
Ensink, J. M. et al. Oral bioavailability and in vitro stability of pivampicillin, bacampicillin, talampicillin, and ampicillin in horses. Am. J. Vet. Res. 57 , 1021–1024 (1996).
Sloane, J. L. et al. Prodrugs of PKC modulators show enhanced HIV latency reversal and an expanded therapeutic window. Proc. Natl Acad. Sci. USA 117 , 10688–10698 (2020).
Citrome, L. Breakthrough drugs for the interface between psychiatry and neurology. Int. J. Clin. Pract. 70 , 298–299 (2016).
Eddy, N. B., Halbach, H. & Braenden, O. J. Synthetic substances with morphine-like effect: clinical experience: potency, side-effects, addiction liability. Bull. World Health Organ. 17 , 569 (1957).
CAS PubMed PubMed Central Google Scholar
Crews, K. R. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for codeine therapy in the context of cytochrome P450 2D6 (CYP2D6) genotype. Clin. Pharmacol. Ther. 91 , 321–326 (2012).
Klous, M. G., Van den Brink, W., Van Ree, J. M. & Beijnen, J. H. Development of pharmaceutical heroin preparations for medical co-prescription to opioid dependent patients. Drug Alcohol Depend. 80 , 283–295 (2005).
Green, K. & DOWNS, S. J. Prednisolone phosphate penetration into and through the cornea. Invest. Ophthalmol. Vis. Sci. 13 , 316–319 (1974).
CAS Google Scholar
Schijvens, A. M., ter Heine, R., de Wildt, S. N. & Schreuder, M. F. Pharmacology and pharmacogenetics of prednisone and prednisolone in patients with nephrotic syndrome. Pediatr. Nephrol. 34 , 389–403 (2019).
Ishikawa, T. Chemotherapy with enteric-coated tegafur/uracil for advanced hepatocellular carcinoma. World J. Gastroenterol. 14 , 2797 (2008).
Diasio, R. B., Bennett, J. E. & Myers, C. E. Mode of action of 5-fluorocytosine. Biochem. Pharmacol. 27 , 703–707 (1978).
Fryklund, J., Gedda, K. & Wallmark, B. Specific labelling of gastric H+,K+-ATPase by omeprazole. Biochem. Pharmacol. 37 , 2543–2549 (1988).
Khan, A. K. A., Piris, J. & Truelove, S. C. An experiment to determine the active therapeutic moiety of sulphasalazine. Lancet 310 , 892–895 (1977).
Article Google Scholar
Meyers, S., Sachar, D. B., Present, D. H. & Janowitz, H. D. Olsalazine sodium in the treatment of ulcerative colitis among patients intolerant of sulfasalazine: a prospective, randomized placebo-controlled, double-blind, dose-ranging clinical trial. Gastroenterology 93 , 1255–1262 (1987).
Green, J. R. B. et al. Balsalazide is more effective and better tolerated than mesalamine in the treatment of acute ulcerative colitis. Gastroenterology 114 , 15–22 (1998).
Gillis, J. C. & Wiseman, L. R. Secnidazole. Drugs 51 , 621–638 (1996).
Kim, I. et al. Identification of a human valacyclovirase: biphenyl hydrolase-like protein as valacyclovir hydrolase. J. Biol. Chem. 278 , 25348–25356 (2003).
Sun, J., Dahan, A. & Amidon, G. L. Enhancing the intestinal absorption of molecules containing the polar guanidino functionality: a double-targeted prodrug approach. J. Med. Chem. 53 , 624–632 (2010).
Wang, T. et al. Discovery of the human immunodeficiency virus type 1 (HIV-1) attachment inhibitor temsavir and its phosphonooxymethyl prodrug fostemsavir. J. Med. Chem. 61 , 6308–6327 (2018).
Kudo, S. & Ishizaki, T. Pharmacokinetics of haloperidol. Clin. Pharmacokinet. 37 , 435–456 (1999).
Daley‐Yates, P. T., Price, A. C., Sisson, J. R., Pereira, A. & Dallow, N. Beclomethasone dipropionate: absolute bioavailability, pharmacokinetics and metabolism following intravenous, oral, intranasal and inhaled administration in man. Br. J. Clin. Pharmacol. 51 , 400–409 (2001).
Chen, K.-J., Plaunt, A. J., Leifer, F. G., Kang, J. Y. & Cipolla, D. Recent advances in prodrug-based nanoparticle therapeutics. Eur. J. Pharm. Biopharm. 165 , 219–243 (2021).
Reker, D. et al. Computationally guided high-throughput design of self-assembling drug nanoparticles. Nat. Nanotechnol. 16 , 725–733 (2021).
Mauro, M. A., Murphy, K. P. J., Thomson, K. R., Venbrux, A. C. & Morgan, R. A. Image-Guided Interventions E-Book. Expert Radiology Series (Elsevier Health Sciences, 2020).
Rosen, L. S. et al. Phase 1 study of TLK286 (Telcyta) administered weekly in advanced malignancies. Clin. Cancer Res. 10 , 3689–3698 (2004).
Gong, J., Yan, J., Forscher, C. & Hendifar, A. Aldoxorubicin: a tumor-targeted doxorubicin conjugate for relapsed or refractory soft tissue sarcomas. Drug Des. Dev. Ther. 12 , 777 (2018).
Sitar, D. S. Clinical pharmacokinetics of bambuterol. Clin. Pharmacokinet. 31 , 246–256 (1996).
Scheeren, T. W. L. Ceftobiprole medocaril in the treatment of hospital-acquired pneumonia. Future Microbiol. 10 , 1913–1928 (2015).
Grygorenko, O. O. et al. Generating multibillion chemical space of readily accessible screening compounds. iScience 23 , 101681 (2020).
Chen, C.-H. in Xenobiotic Metabolic Enzymes: Bioactivation and Antioxidant Defense 155–168 (Springer, 2020).
Beverage, J. N., Sissung, T. M., Sion, A. M., Danesi, R. & g, W. D. CYP2D6 polymorphisms and the impact on tamoxifen therapy. J. Pharm. Sci. 96 , 2224–2231 (2007).
Krzyszczyk, P. et al. The growing role of precision and personalized medicine for cancer treatment. Technology 6 , 79–100 (2018).
Baird, J. K. 8-Aminoquinoline therapy for latent malaria. Clin. Microbiol. Rev. 32 , e00011-19 (2019).
Inturrisi, C. E. et al. Evidence from opiate binding studies that heroin acts through its metabolites. Life Sci. 33 , 773–776 (1983).
Schmitt-Hoffmann, A. et al. Single-ascending-dose pharmacokinetics and safety of the novel broad-spectrum antifungal triazole BAL4815 after intravenous infusions (50, 100, and 200 milligrams) and oral administrations (100, 200, and 400 milligrams) of its prodrug, BAL8557, in healthy volunteers. Antimicrob. Agents Chemother. 50 , 279–285 (2006).
Mackman, R. L. Phosphoramidate prodrugs continue to deliver, the journey of remdesivir (GS-5734) from RSV to SARS-CoV-2. ACS Med. Chem. Lett. 13 , 338–347 (2022).
Jordan, V. C. Tamoxifen: a most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2 , 205–213 (2003).
Jordan, V. C., Collins, M. M., Rowsby, L. & Prestwich, G. A monohydroxylated metabolite of tamoxifen with potent antioestrogenic activity. J. Endocrinol. 75 , 305–316 (1977).
Mickle, T., Guenther, S. & Chi, G. Methylphenidate-prodrugs, processes of making and using the same. US patent 10584112-B2 (2020).
O’Neill, J. I. M. Antimicrobial resistance: tackling a crisis for the health and wealth of nations. Review on Antimicrobial Resistance https://amr-review.org/sites/default/files/AMR%20Review%20Paper%20-%20Tackling%20a%20crisis%20for%20the%20health%20and%20wealth%20of%20nations_1.pdf (2014).
Evans, L. E. et al. Exploitation of antibiotic resistance as a novel drug target: development of a β-lactamase-activated antibacterial prodrug. J. Med. Chem. 62 , 4411–4425 (2019).
Caradec, T. et al. A novel natural siderophore antibiotic conjugate reveals a chemical approach to macromolecule coupling. ACS Cent. Sci. 9 , 2138–2149 (2023).
Peukert, C. et al. Siderophore conjugation with cleavable linkers boosts the potency of RNA polymerase inhibitors against multidrug-resistant E. coli . Chem. Sci. 14 , 5490–5502 (2023).
Bodor, N., Shek, E. & Higuchi, T. Delivery of a quaternary pyridinium salt across the blood-brain barrier by its dihydropyridine derivative. Science 190 , 155–156 (1975).
Bodor, N. & Simpkins, J. W. Redox delivery system for brain-specific, sustained release of dopamine. Science 221 , 65–67 (1983).
Peauger, L. et al. Donepezil-based central acetylcholinesterase inhibitors by means of a “bio-oxidizable” prodrug strategy: design, synthesis, and in vitro biological evaluation. J. Med. Chem. 60 , 5909–5926 (2017).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7 , 42717 (2017).
Fralish, Z., Chen, A., Skaluba, P. & Reker, D. DeepDelta: predicting ADMET improvements of molecular derivatives with deep learning. J. Cheminform 15 , 101 (2023).
Reker, D., Hoyt, E. A., Bernardes, G. J. L. & Rodrigues, T. Adaptive optimization of chemical reactions with minimal experimental information. Cell Rep. Phys. Sci. 1 , 100247 (2020).
Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5 , 1572–1583 (2019).
Coley, C. W. et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 365 , eaax1566 (2019).
Zaretzki, J., Matlock, M. & Swamidass, S. J. XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 53 , 3373–3383 (2013).
de Bruyn Kops, C. et al. GLORY: generator of the structures of likely cytochrome P450 metabolites based on predicted sites of metabolism. Front. Chem. 7 , 402 (2019).
Olsen, L., Montefiori, M., Tran, K. P. & Jørgensen, F. S. SMARTCyp 3.0: enhanced cytochrome P450 site-of-metabolism prediction server. Bioinformatics 35 , 3174–3175 (2019).
Wishart, D. S. et al. BioTransformer 3.0—a web server for accurately predicting metabolic transformation products. Nucleic Acids Res. 50 , W115–W123 (2022).
Šícho, M. et al. FAME 3: predicting the sites of metabolism in synthetic compounds and natural products for phase 1 and phase 2 metabolic enzymes. J. Chem. Inf. Model. 59 , 3400–3412 (2019).
Karaman, R., Dajani, K. K., Qtait, A. & Khamis, M. Prodrugs of acyclovir–a computational approach. Chem. Biol. Drug Des. 79 , 819–834 (2012).
Markovic, M., Ben-Shabat, S. & Dahan, A. Computational simulations to guide enzyme-mediated prodrug activation. Int. J. Mol. Sci. 21 , 3621 (2020).
Kim, S. et al. PubChem substance and compound databases. Nucleic Acids Res. 44 , D1202–D1213 (2016).
Elia, J., Easley, C. & Kirkpatrick, P. Lisdexamfetamine dimesylate. Nat. Rev. Drug Discov. 6 , 343–344 (2007).
Hung, A., Sinclair, M., Hemmersbach-Miller, M., Edmonston, D. & Wyatt, C. Prescribing rates and characteristics of recipients of tenofovir-containing regimens before and after market entry of tenofovir alafenamide. J. Manag. Care Spec. Pharm. 26 , 1582–1588 (2020).
PubMed Google Scholar
D’Angelo, A. B., Westmoreland, D. A., Carneiro, P. B., Johnson, J. & Grov, C. Why are patients switching from tenofovir disoproxil fumarate/emtricitabine (Truvada) to tenofovir alafenamide/emtricitabine (Descovy) for pre-exposure prophylaxis? AIDS Patient Care STDS 35 , 327–334 (2021).
Urquhart, L. Top product forecasts for 2020. Nat. Rev. Drug Discov. 19 , 86–87 (2020).
Urquhart, L. Top companies and drugs by sales in 2020. Nat. Rev. Drug Discov. 20 , 253 (2021).
Urquhart, L. Top companies and drugs by sales in 2021. Nat. Rev. Drug Discov. 21 , 251 (2022).
Urquhart, L. Top companies and drugs by sales in 2022. Nat. Rev. Drug Discov. 22 , 260 (2023).
Cha, M. & Yu, F. Pharma’s first-to-market advantage. https://www.mckinsey.com/~/media/McKinsey/Industries/Pharmaceuticals%20and%20Medical%20Products/Our%20Insights/Pharmas%20first%20to%20market%20advantage/Pharmas%20first%20to%20market%20advantage.pdf (McKinsey & Co., 2014).
Orayj, K. & Lane, E. Patterns and determinants of prescribing for Parkinson’s disease: a systematic literature review. Parkinsons Dis. 2019 , 9237181 (2019).
PubMed PubMed Central Google Scholar
Download references
Acknowledgements
Z.F. is supported by the Department of Defense (DoD) through the National Defense Science & Engineering Graduate (NDSEG) Fellowship Program. P.Z. acknowledges the support of NIAID grants AI139216 and AI152896. D.R. acknowledges the support of NIGMS grant R35GM151255.
Author information
Authors and affiliations.
Department of Biomedical Engineering, Duke University, Durham, NC, USA
Zachary Fralish & Daniel Reker
Department of Computer Science, Duke University, Durham, NC, USA
Ashley Chen
Rivus Pharmaceuticals, Charlottesville, VA, USA
Shaharyar Khan
Department of Biochemistry, Duke University School of Medicine, Durham, NC, USA
You can also search for this author in PubMed Google Scholar
Contributions
All authors made a substantial, direct and intellectual contribution to the work and approved it for publication.
Corresponding author
Correspondence to Daniel Reker .
Ethics declarations
Competing interests.
D.R. acts as a consultant to the pharmaceutical and biotechnology industry, as a mentor for Start2, and serves on the scientific advisory board of Areteia Therapeutics. S.K. is Chief Scientific Officer at Rivus. P.Z. is a scientific co-founder of Valanbio Therapeutics aiming at developing novel antibiotics.
Peer review
Peer review information.
Nature Reviews Drug Discovery thanks Jarkko Rautio and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
ChEBI Database: https://www.ebi.ac.uk/chebi/
ClinCalc DrugStats Database: https://clincalc.com/DrugStats/
DrugBank: https://go.drugbank.com/
PubChem: https://pubchem.ncbi.nlm.nih.gov/
Therapeutic Target Database: https://idrblab.net/ttd/
Supplementary information
Supplementary table 1, supplementary table 2, supplementary table 3, supplementary table 4, supplementary table 5, supplementary table 6, supplementary table 7, supplementary table 8, supplementary table 9, supplementary table 10, supplementary table 11, supplementary table 12, rights and permissions.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Cite this article.
Fralish, Z., Chen, A., Khan, S. et al. The landscape of small-molecule prodrugs. Nat Rev Drug Discov 23 , 365–380 (2024). https://doi.org/10.1038/s41573-024-00914-7
Download citation
Accepted : 16 February 2024
Published : 02 April 2024
Issue Date : May 2024
DOI : https://doi.org/10.1038/s41573-024-00914-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

Advertisement

- Previous Article
- Next Article
Introduction
Approaches to deliver genes encoding prodrug-activating enzymes into tumor cells, approaches to deliver prodrug-activating enzymes into tumor cells or tissues, which approach is better: gdept, vdept, or adept, future perspective, strategies for enzyme/prodrug cancer therapy 1.
To whom requests for reprints should be addressed, at Washington University Medical School, Campus Box 8069, 660 South Euclid Avenue, St. Louis, MO 63110. Phone: (314) 747-5186; Fax: (314) 747-2797; E-mail: [email protected]
- Split-Screen
- Article contents
- Figures & tables
- Supplementary Data
- Peer Review
- Open the PDF for in another window
- Get Permissions
- Cite Icon Cite
- Search Site
- Version of Record November 1 2001
Guang Xu , Howard L. McLeod; Strategies for Enzyme/Prodrug Cancer Therapy 1 . Clin Cancer Res 1 November 2001; 7 (11): 3314–3324.
Download citation file:
- Ris (Zotero)
- Reference Manager
The selective activation of prodrug(s) in tumor tissues by exogenous enzyme(s) for cancer therapy can be accomplished by several ways, including gene-directed enzyme prodrug therapy (GDEPT), virus-directed enzyme prodrug therapy (VDEPT), and antibody-directed enzyme prodrug therapy (ADEPT). The central part of enzyme/prodrug cancer therapy is to deliver drug-activating enzyme gene or functional protein to tumor tissues, followed by systemic administration of a prodrug. Although each approach (GDEPT, VDEPT, and ADEPT) has been tested in clinical trials, there are some potential problems using the current delivery systems. In this article, disadvantages and advantages associated with each approach (GDEPT, VDEPT, and ADEPT) and future perspective for improving current systems are discussed.
Chemotherapy is an important treatment for cancer patients. However, its success is limited by several drawbacks, including insufficient drug concentrations in tumors, systemic toxicity, lack of selectivity for tumor cells over normal cells, and the appearance of drug-resistant tumor cells (1, 2, 3, 4, 5, 6, 7) . A number of strategies have been used to overcome these problems, including alternative formulations ( e.g. , liposomes; Ref. 8 ), resistance modulation ( e.g. , PSC833; Ref. 9 ), antidotes/toxicity modifiers ( e.g. , ICRF-187; Ref. 10 ), and gene therapy. One promising area for improving tumor selectivity is enzyme prodrug therapy.
Enzyme-activating prodrug therapy is a two-step approach. In the first step, a drug-activating enzyme is targeted and expressed in tumors. In the second step, a nontoxic prodrug, a substrate of the exogenous enzyme that is now expressed in tumors, is administered systemically (2, 3, 4) . The net gain is that a systemically administered prodrug can be converted to high local concentration of an active anticancer drug in tumors. To be clinically successful, both enzymes and prodrugs should meet certain requirements for this strategy. The enzymes should be either of nonhuman origin or human protein that is absent or expressed only at low concentrations in normal tissues (11, 12) . The protein must achieve sufficient expression in the tumors and have high catalytic activity (13) . The prodrug should be a good substrate for the expressed enzyme in tumors but not be activated by endogenous enzyme in nontumor tissues. It must be able to cross the tumor cell membrane for intracellular activation, and the cytotoxicity differential between the prodrug and its corresponding active drug should be as high as possible. It is preferred that the activated drug be highly diffusible or be actively taken up by adjacent nonexpressing cancer cells for a “bystander” killing effect, the ability to kill any neighboring nonexpressing cells (13) . In addition, the half-life of active drug should be long enough to induce a bystander effect but short enough to avoid the drug leaking out into the systemic circulation (13) .
Currently, delivery methods for an enzyme/prodrug strategy can be divided into two major classes: ( a ) delivery of genes that encode prodrug-activating enzymes into tumor tissues (GDEPT, 3 VDEPT, etc.); and ( b ) delivery of active enzymes onto tumor tissues (ADEPT). An overview of these strategies is shown in Fig. 1 . The aim of this review is to summarize some of the areas of recent progress in enzyme-activating prodrug therapy and discuss areas of future development.
GDEPT, also known as suicide gene therapy, is a technique that involves physical delivery of a gene for a foreign enzyme to tumor cells where a systemically administered nontoxic prodrug can be activated after expression of the enzyme (5, 13, 14, 15, 16) . Many GDEPT studies have used liposomal gene delivery, but the challenge of vector delivery is common for all areas of gene therapy and has been exhaustively reviewed elsewhere (3, 11, 13) . An early example of GDEPT is the combination of HSV-TK and GCV. GCV is an antiviral drug, which is phosphorylated by HSV-TK and then by cellular kinases to produce GCV triphosphate, which disrupts DNA synthesis during S phase, leading to cell death (1, 4, 16, 17, 18, 19, 20) . A second early example is the combination of the bacterial CD and the antifungal drug 5-FC, which was effective to kill tumor cells after the conversion by CD to active 5-FU (20, 21) . In addition to the above classical examples, many other enzyme/prodrug combinations have been used for GDEPT ( Table 1 ; Refs. 13 and 22 ).
Many of the enzyme/prodrug systems applied in GDEPT, including HSV-TK/GCV, Escherichia coli CD/5-FC, and E. coli NTR/CB1954, act intracellularly by converting prodrugs into active drugs within cells (2) . This mode of action requires cell-cell contact to mediate maximal killing. Recently, an extracellular cytotoxic effector system was reported in which a secreted form of lysosomal human β-glucuronidase, which converts an inactive glucuronidated derivative of doxorubicin (HMR 1826) to the cytotoxic doxorubicin, was introduced into tumor cells (2) . There was evidence that a bystander effect resulted in a significant tumor cell killing both in vitro and in a human xenograft model in nude mice (2) . The advantage of this extracellular system is that a hydrophilic prodrug was converted into a lipophilic, cell-permeable cytotoxic drug outside cells so that both transduced and nontransduced cells were targeted. As cell-cell contact is not required for a bystander effect, the killing ability of this system is enhanced. To explore the mechanism of a bystander effect of the NTR/CB1954 system, hydroxylamine metabolites of CB1954 were found to be able to cross cell membranes freely and are responsible for a bystander effect, independent of gap junctions (23) . Although the cell-permeable active metabolites of prodrugs for GDEPT have advantages in certain situations, such as tumors with few gap junctions, the downside of this mode is the cytotoxicity to adjacent normal tissues and the risk of systemic diffusion (23) .
GDEPT can also be used to improve the selectivity of currently used agents. CYP-based prodrug activation systems is one example that shows promise for clinical use (27) . Members of the CYP enzyme superfamily convert the chemotherapeutic agents cyclophosphamide and IFA to active alkylating agents that cause cell death (27) . The expression of CYP is generally high in liver but lower in tumor cells (27) , providing a potential mechanism for intrinsic drug resistance. Tumor cells are highly sensitive to cyclophosphamide or IFA after the delivery of the CYP2B1 gene into tumor cells both in vitro and in vivo (28) . To additionally improve and increase the efficiency of this system, a two enzymes/one prodrug system was developed. In this study, CYP2B1 was coexpressed with the RED gene and used to activate prodrug cyclophosphamide in tumor cells (18) . The rationale is that CYP-catalyzed prodrug activation is dependent on electron reduction from the flavoenzyme RED, an enzyme widely expressed in many cell types, including tumor cells. Despite the significant level of endogenous basal expression, the authors found that RED gene transfer greatly enhanced the cyclophosphamide sensitivity of tumor cells transfected with CYP2B1. Thus, endogenous RED levels in tumor tissues may be an important determinant for the sensitivity of tumor cells to CYP/cyclophosphamide gene therapy. This result also suggested that CYP-based cancer gene therapy might be particularly effective for tumors that express a high endogenous level of RED.
Recently, an approach to improve the CPA/MTX-α-peptide system was described (4) . CPA is a zymogen that becomes catalytically active after its propeptide is removed by trypsin (4) . Activated CPA converts MTX-α-peptide prodrug into active MTX that inhibits dihydrofolate reductase and causes cell death. Because trypsin is localized in the small intestine but is absent in tumors, the prodrug activation by CPA is limited to the intestine, causing local toxicity and low drug concentration in tumors. To activate the prodrug intratumorally in a trypsin-independent manner, a battery of CPA mutants were created in which the trypsin cleavage sites were mutated at the recognition sites for mammalian propeptidases. These constructs were cotransfected into COS-1 cells with propeptidases, endogenous cellular enzymes present in tumor cells. They found that one mutant, CPA95, had a high level of endogenously activated enzyme in cotransfected cells, and it was able to secrete and sensitize tumor cells to MTX-α-peptide in a manner similar to that of trypsin-activated wild-type CPA (4) . This mutant CPA/MTX prodrug system might represent a novel approach in activating prodrug intratumorally attributable to bypassing an activation step. An additional advantage of this system is the potential use for other antifolates, such as the α-peptide conjugates of thymidylate synthase inhibitors.
VDEPT is a pharmacologically oriented gene therapy strategy that uses viral vectors to deliver a gene that encodes an enzyme that is capable of converting a systemically administrated nontoxic prodrug into a cytotoxic agent within tumor cells (20, 29) . The NTR/CB1954 combination was an initial example of VDEPT in which colorectal and pancreatic cancer cells were found to be sensitized to CB1954 after retroviral transduction and expression of the E. coli NTR gene (20, 30) . Currently, several viruses have been used for VDEPT, including retroviruses, adenoviruses, HSV (31) , adeno-associated virus (32, 33, 34) , lentivirus, and EBV (35) . Over the years, many drug-activating enzyme gene/prodrug combinations have been delivered into tumors in vitro or in vivo by VDEPT, the majority using CD/5-FC or HSV-TK/GCV with the involvement of retroviral and adenoviral vectors. These examples were reviewed elsewhere (29, 36) . Several recent illustrations of VDEPT are described below and are also listed in Table 2 to highlight novel therapeutic strategies.
Recently, recombinant retroviruses were used to individually deliver six different cyclophosphamide- or IFA-metabolizing human CYP genes to 9 L gliosarcoma cells (43) . It was found that CYP2B6 and CYP2C18 transfection yielded pronounced cytotoxicity after cyclophosphamide treatment, with more efficient prodrug activation and cytotoxicity observed after cotransfection with RED. Antitumor activity was also seen after VDEPT in immunodeficient mice bearing tumor xenografts. Taken together, the results indicated that either CYP2B6 or CYP2C18 plus RED may be an excellent combination for use with prodrug cyclophosphamide in CYP-based cancer gene therapy. A retroviral gene transfer approach has also been used for NTR/CB1954 enzyme-prodrug therapy by two different groups (20, 30) . In female BALB/c nude mice bearing s.c. NTR-expressing human pancreatic carcinoma cells, administration of CB1954 resulted in tumor regression, growth delay, and significantly increased median survival (20) . In the other study, the E. coli NTR gene was introduced into colorectal and pancreatic cancer cell lines by retroviral delivery. It was shown that NTR-expressing clones of both cell lines were more sensitive to cytotoxic effects mediated by the prodrug CB1954 (30) , suggesting that NTR and CB1954 may be an attractive combination for VDEPT.
NTR has also been delivered into tumor cells by a replication-defective adenovirus vector containing an NTR expression cassette (40) . In this study, ovarian carcinoma cells were grown at low-serum condition, mimicking the low rate of cell proliferation in human tumors. It was shown that NTR-expressing cells were sensitive to CB1954. Additionally, cisplatin-resistant cells that expressed NTR were also susceptible to CB1954, suggesting this system might be useful for patients with cisplatin-resistant tumors. An adenoviral vector was used to carry the human CES gene, driven by the cytomegalovirus promoter, to infect human lung adenocarcinoma cells in vitro and in vivo (39) . It was shown that CES was efficiently expressed in these cells. In the presence of CPT-11, the growth of adenovirus-infected cells was greatly inhibited, and additional cell mixing studies indicated the presence of a bystander effect in this system. Results from in vivo studies using nude mice bearing s.c. A549 tumors and receiving CPT-11 at three sites within the tumor indicated that there was a significant reduction in tumor size.
Besides the above in vitro viral delivery systems, either retroviral or adenoviral vectors have been used to deliver HSV-TK into human tumors in Phase I/II clinical trials (44, 45, 46, 47, 48) . The targeted human tumors included recurrent glioblastoma multiforme (44, 46) , adenocarcinoma of the prostate (45) , and malignant mesothelioma (37) , e.g. , 20 patients with untreated mesothelioma in the pleural cavity were selected as a model for treatment of a localized malignancy (37) . Adenoviral vector-mediated intrapleural HSV-TK injection (1 × 10 9 –1 × 10 12 plaque-forming units), followed by systemic administration of GCV for 2 weeks, was given to these patients. HSV-TK gene transfer was observed in 11 of 20 patients in a dose-related manner, with minimal side effects, suggesting treatment of localized malignancy using this vector is feasible. No evidence of clinical activity was noted in this study (37) . The main clinical challenge appears to be overcoming the failure to achieve cDNA expression in a sufficient number of human tumor cells.
Despite extensive use of retroviral and adenoviral vectors to deliver prodrug-activating enzyme genes, both vectors have some disadvantages, which limit the use of VDEPT (42) . The major disadvantage associated with a retroviral vector is that recombinant retroviruses only target dividing cells, whereas most human tumor cells are slowly dividing, yielding a low transduction rate (2–10%; Ref. 42 ). When this strategy uses HSV-TK/GCV or CD/5-FC to generate antimetabolites, which also require cell division for activity, it is not surprising that the results are less than dramatic. However, this drawback could be beneficial in some case. Brain tumors, where only tumor cells are proliferating, allow for a high tumor:normal transfection differential for retroviral delivery (48) . The low retrovirus titer, leading to decreased infection efficiency, is another drawback. Some researchers have been trying to increase retrovirus titer for VDEPT. It was found that prolonged low speed centrifugation during viral preparation was a simple way to concentrate recombinant retrovirus ∼100-fold (20) . Another disadvantage is that retroviruses produced from murine or dog cells are all sensitive to human serum when applied in human subjects, whereas viral particles generated from human cells are more resistant (13) . To solve this problem, efforts have been made to develop a variety of packaging cell lines that produce high-titer recombinant retroviruses resistant to human serum. In addition, a soluble protein called Galα1–3Gal was found to protect retroviruses from human serum when coadministered with retroviruses (13) . The other disadvantages associated with retroviral vectors include immunogenicity, risk of insertional mutagenesis, risk of reversal to wild-type virus, envelope-induced complement-mediated inactivation, difficulties in producing high-titer viruses, and only targeting dividing cells (11, 13) . Compared with retroviruses, adenoviruses have some advantages, including higher titers capable of generating infections in both dividing and nondividing cells (13) . The disadvantages of adenoviral vectors include immunogenenicity, reversal to wild type, and short periods of gene expression in dividing cells (11) . In addition to adenoviral and retroviral vectors, an EBV-based viral vector has been used to deliver exogenous enzyme-encoding CD or NTR into EBV-positive B-cell lines to activate 5-FC or CB1954, respectively (35) . Both enzyme systems were effective to kill tumor cells in vitro in a prodrug-dependent manner.
Both the adenoviral and retroviral vectors mentioned above are replication incompetent, which limit their ability to infect additional cells subsequent to the initial infection event (49) . Although the bystander effect increases the degree of tumor cell killing, additional strategies are needed to enhance the therapeutic efficacy of VDEPT. Viral prodrug-activating enzyme constructs have been engineered to retain viral oncolytic potential and replication capability. The most well-studied example is Onyx-015, an E1B-55D-deleted adenovirus that selectively replicates and lyses p53-deficient cancer cells, while sparing cells with functional p53 (50, 51, 52, 53) . Over 10 clinical trials (Phase I–III) in >200 patients with recurrent squamous cell carcinoma of the head and neck indicated that this single viral agent alone has antitumor activity (50, 54, 55) . There is a suggestion that this activity is additionally enhanced when Onyx-015 is combined with conventional cytotoxic chemotherapy (51) . Onyx-015 has been used to express HSV-TK for the treatment of human colon cancer xenografts in nude mice (49) . E1B-55D-deleted adenovirus carrying HSV-TK alone was as effective as a standard replication-deficient adenoviral vector expressing HSV-TK in combination with GCV. The addition of GCV enhanced the antitumor effect of E1B-55D-deleted adenovirus carrying HSV-TK by 5-fold (49) . Recently, combined oncolysis and prodrug bioactivation was studied using a HSV-1 mutant in which the viral ribonucleotide reductase gene was inactivated by insertion of transgene sequences encoding yeast CD (31) . In cultured human colon carcinoma cells, the virus effectively destroyed cells by oncolysis and simultaneously induced conversion of the prodrug 5-FC to 5-FU by yeast CD, resulting in enhanced cytotoxicity when compared with either approach alone (31) . The similar results were also observed in mice bearing colon cancer xenografts. The combination of direct viral oncolysis and suicide gene system appears to offer an advantage over either approach alone and may represent an important future direction for the integration of VDEPT into clinical practice.
GPAT is a variation of GDEPT, which uses known transcriptional differences between normal and tumor cells to drive the selective expression of a drug-metabolizing enzyme to convert a nontoxic prodrug into a toxic moiety. TREs are placed upstream of the enzyme gene, driving selective expression (11, 13, 56) . A number of tumor-specific TREs have been used, including genes that are amplified in tumor cells compared with normal cells or genes that express tumor-associated antigens, such as CEA for colorectal cancer or N-myc for neuroblastoma. Alternatively, TREs of tissue-specific genes can also be used for GPAT (Refs. 11 and 56, 57, 58 ; Table 3 ).
The goal of GPAT is tumor-selective therapy, and this strategy has been used for breast and pancreatic cancer therapy, using TREs of the MUC1 and erb B2 genes. The level of MUC1 gene product, the polymorphic epithelial mucin, is increased in most carcinomas, and the level of the erb B2 product is overexpressed in breast and pancreatic cancers (57) . The TRE of the MUC1 or erb B2 gene was placed upstream of the HSV-TK gene, and this expression cassette was placed in a retroviral vector (57) . Transduction with retroviruses containing only the MUC1 promoter and HSV-TK gene increased GCV sensitivity in MUC1-positive cells. The cytotoxic effect was additionally enhanced by transduction with retroviruses containing chimeric MUC1/ erb B2 promoters and HSV-TK gene, suggesting a useful role of multiple TREs to drive expression of the suicide gene. In a recent study, the ODC promoter was used to up-regulate expression of the rabbit CES, a drug-activating enzyme for CPT-11, in N-myc overexpressing neuroblastoma cells (Ref. 59 ; Fig. 2 ). This strategy was based on the fact that MYC family proteins (c-, N-, and L-) are activators of the ODC promoter by dimerizing with the transcription factor MAX (59) . Introducing the ODC/CES cDNA cassette into N-myc overexpressing neuroblastoma cells gave increased CES activity in situ , and tumor cells were 10-fold more sensitive to the prodrug CPT-11. This suggests that the MYC/ODC/CES and prodrug CPT-11 may be a good candidate for GPAT of neuroblastoma, glioblastoma, and lung or colon carcinomas, because MYC family transcription factors are overexpressed in these tumors.
GPAT has been applied clinically. Thus far, there are ∼20 Phase I trials for GPAT in progress worldwide (58) . HSV-TK and CD systems are the main activating enzymes used in these clinical trials. The first targeted gene therapy trial for breast cancer and the first to use the CD system in human subjects were performed by Panhda et al. (56) . Because overexpression of erb B2 was found in 20–50% of breast carcinoma, the erb B2 promoter was used to drive the tumor-specific expression of the E. coli CD gene (56) . In a Phase I clinical trial, the therapy was performed in 12 breast cancer patients who received both intratumoral injections of a plasmid construct containing the therapeutic cassette of the E. coli CD gene driven by the erb B2 promoter and systemic administration of the prodrug 5-FC. The expression of the therapeutic construct was observed in the majority of injected nodules, with expression limited to erb B2-positive tumor cells, and no expression was detected in adjacent normal tissues, indicating an excellent tumor selectivity (56) . In addition, there was a reduction in tumor size after plasmid injection and systemic prodrug administration in 4 of 12 patients (33%), without causing local or systemic complications (56) . Results such as these provide additional encouragement for the development of GPAT strategies.
Other Genetic Approaches.
In addition to the above approaches, genetically modified cells have been used to express drug-activating enzyme genes in tumors. In this approach, drug-activating enzyme genes are stably transfected into cells that are additionally encapsulated by cellulose sulfate. The engineered cells are then introduced into tumors by injection in an immunoprotected environment to produce enzymes in tumors (7) . This method was developed as a novel approach that combines gene/cell therapy with chemotherapy (7) . It was also considered as a safe and easy application for clinical use, because delivery of suicide gene-transfected/encapsulated cells is a feasible clinical approach without involving direct gene therapeutic interference in patients.
Using this approach, CYP2B1 was delivered into mice for tumor therapy (7, 60) . In these studies, encapsulated human embryonal kidney epithelial 293 cells expressing CYP2B1, under the control of the cytomegalovirus immediate early promoter, were administered into mice by two routes: ( a ) to deliver the capsules directly into the tumors in nude mice (7) ; and ( b ) to implant capsules adjacent to pre-established pancreatic tumors in nude mice (60) . Low doses of the prodrug IFA were administered to tumor-bearing mice every 3rd day for 2 weeks in both studies. Tumor regression was achieved after 3 weeks, with no tissue reaction or pancreatitis observed 7 days after injection (7, 60) . A similar result was observed when Feline kidney cells were used for CYP2B1 expression (7) . Human breast cancer cells (MDA-MB-361) have also been used to express enzymatically active surface-tethered bacterial CPG2(Q)-3 (61) . In this study, engineered breast cancer cells were mixed with nonexpressing cells, and the resultant mixtures were injected into nude mice that had a breast carcinoma xenograft. After 4 days, the prodrug CMDA was administrated into those mice. Expression of the drug-activating enzyme was able to convert the prodrug into the cytotoxic moiety in vivo , resulting in either cures or tumor regression in all surface-tethered CPG2(Q)-3-expressing groups (61) . Furthermore, CPG2 activity was not detected in blood samples, indicating there was no significant shedding of the enzyme into the blood circulation, and high level of selectivity for the surface-tethered approach was achieved.
ADEPT is a strategy in which a tumor-associated monoclonal antibody is linked to a drug-activating enzyme to create a systemically administered conjugate that only targets tumor tissues. Nontoxic prodrug is then administrated systemically and is converted by the pretargeted enzyme localized on the tumor surface into a toxic drug, resulting in cytotoxic effects in tumor cells (6, 62, 63, 64, 65, 66, 67) . The ideal drugs for ADEPT are small molecules that can diffuse within the tumor tissues, including both antigen-positive and antigen-negative tumor cells, and cause a bystander effect (62, 63, 64) . When ADEPT is applied clinically, the interval between enzyme and prodrug administrations should be optimized so that the conjugate is only accumulated in tumors rather than in blood and normal tissues, to avoid systemic toxicity. ADEPT has been used to deliver many drug-activating enzyme genes to tumors in vitro and in vivo , and recent examples are described below and are also listed in Table 4 .
There are a number of general considerations for ADEPT. The target antigen should be either expressed on the tumor cell membrane or secreted into the extracellular matrix of the tumor (69) , and the use of a high affinity monoclonal antibody is essential (62) . The enzyme should be able to exert its optimal activity at a pH close to that of the tumor extracellular fluid. Because antibody-enzyme conjugate may be immunogenic, circulating host anticonjugate antibodies may interfere with treatment. Therefore, the drug chosen should be dose dependent and cell cycle independent (62) . Ideally, the enzyme system should not have a human homologue to avoid prodrug activation outside the tumor site (64) .
Because the interval between enzyme and prodrug administrations is important for ADEPT, some studies were performed to explore the optimal interval in animals. Linking the enzyme CPG2 to the anti-CEA antibody A5B7, a rapid clearance of conjugate from the circulation was demonstrated, allowing the prodrug CMDA to be safely given 48 h or 72 h after antibody-enzyme administration (63) . In human subjects, 7 days were needed for adequate clearance of antibody-enzyme conjugate from the plasma before the prodrug may be administrated safely, to avoid activation of prodrug in plasma and subsequent systemic toxicity (62) . Recently, this CMDA/CPG2 prodrug/enzyme system has been used in a Phase I clinical trial of 10 patients with colorectal carcinoma (70) . The bacterial enzyme CPG2 was conjugated to the F(ab′) 2 fragment of murine A5B7 monoclonal Ab, and a galactosylated second clearing Ab against CPG2 was also used to lower levels of conjugate in the circulation and other nontumor tissues. The aim of the trial was to measure plasma levels of the prodrug CMDA and active drug CJS11, a bifunctional alkylating agent, released from prodrug by the action of CPG2 localized in tumors. CPG2 activity was found in metastatic tumor biopsies, but not found in normal tissues, after applying the clearing agent. The rapid appearance of the active drug with half-life of 36 ± 14 min in plasma was observed in this system (70) . This initial example provides promise for the use of ADEPT to improve the selectivity of current therapy for solid tumors.
Like GDEPT and VDEPT, there are many clinical limitations associated with ADEPT. In poorly vascularized tumors, delivery of the large conjugate is restricted, and it is not possible to deliver antibody/enzyme conjugate to all of the tumor cells (5, 70) . Because the enzyme level is low, it is very difficult to generate adequate quantities of active drug to reach the lethal concentration. Furthermore, the binding of the conjugate to the cell surface is limited by antigen heterogeneity. Other drawbacks of ADEPT include cost and difficulties with development and purification of antibodies, immunogenicity of antibodies, accessibility of tumor to the enzyme/antibody conjugate, and the conversion of prodrugs in nontumor tissues (69) . The main problem with ADEPT is the immunogenicity of the antibody-enzyme conjugate, which limits multiple cycles of its application (5, 62) . To solve this problem, several solutions have been tried, including the use of humanized proteins and concomitant administration of immunosuppression (62) .
Because of the problems mentioned above, many ways have been tried to improve ADEPT. The first way to improve ADEPT is to use a three-phase system to speed up the removal of enzymes from the circulation without affecting the enzyme activity in tumor tissues (62, 66) . In this approach, a galactosylated anticonjugate antibody was applied after the administration of conjugate and prodrug as a clearing agent that reacted with the conjugate in the plasma, thus decreasing its blood levels, but retaining enzymatic activity in tumors (62) . A second way to improve ADEPT is to use a conjugate containing an enzyme and a partial fragment of antibody, which would be cleared more rapidly from the circulation, with the prodrug given earlier, whereas the enzyme level within the tumor is at the peak concentrations (67) . The third way to improve ADEPT is to combine ADEPT with an antivascular agent, a drug that selectively inhibits tumor blood flow and causes extensive necrosis. In a study of nude mice bearing a colorectal tumor xenograft, a conjugate containing the bacterial CPG2 and the F(ab′) 2 fragment of anti-CEA antibody to activate the prodrug CMDA was combined with the antivascular agent 5, 6-dimethylxanthenone-4-acetic acid at 20 h postconjugate injection, resulting in killing a larger part of tumor, doubling the concentration of antibody-enzyme conjugate retained in tumor, and significantly prolonging the tumor growth inhibition caused by ADEPT alone (66) . Furthermore, 5, 6-dimethylxanthenone-4-acetic acid also increased prodrug retention within the tumor by 16-fold. The fourth way to improve ADEPT is to use mutant form(s) of human enzymes to avoid systemic toxicity caused by the use of wild-type human enzyme(s) and decrease immune responses caused by the use of nonhuman enzyme(s) (6) . A mutant form of human CPA conjugated to a tumor-associated antibody was effective to activate several prodrugs, including thymidylate synthase inhibitors GW 1031 and GW 1843 and the dihydrofolate reductase inhibitor MTX, whereas all these prodrugs were not efficient substrates for endogenous CPA. The use of mutant human enzymes may provide less immunogenicity than nonendogenous enzymes and less systemic toxicity than endogenous enzymes (6) . The final way to improve ADEPT is to use recombinant DNA technology to produce a fusion protein with defined characteristics and to avoid additional antibody purification steps, which may cause reduced enzymatic activity or decreased antibody binding of the conjugate (67) . An antibody-human β-glucuronidase fusion protein was studied in mice bearing a human tumor xenograft and found to be rapidly cleared from the blood, with a tumor: blood ratio > 100:1 at 7 days after injection. The combination of this fusion protein with a doxorubicin prodrug resulted in superior growth inhibition when compared with prodrug alone (68) . Recently, an expression plasmid for the production of a fusion protein containing the single-chain Fv anti-CD20 mouse monoclonal antibody and human lysosomal enzyme β-glucuronidase was found to bind CD20-expressing lymphoma cells in a specific manner and was able to activate the prodrug N -[4-daunorubicin- N -carbonyl (poxymethyl)phenyl] O -b-glucuronyl carbamate at a rate similar to that of purified human β-glucuronidase (67) .
All three enzymatic-prodrug strategies have practical advantages for optimizing the treatment of human cancer. GDEPT and VDEPT have an advantage over ADEPT in that most enzymes need cofactor(s) that is present only inside the cells. Therefore, enzymes delivered by ADEPT may need to gain access inside the cells before they can optimally activate prodrugs. This requirement is limited by the poor penetration of large-sized antibody-enzyme conjugates. In GDEPT, gene-encoding enzymes can be specifically delivered to target tissues by the use of tissue-specific elements, to drive the expression of the enzyme within the target cells (60) . Despite this idea, some theoretical risks for GDEPT, including insertional mutagenesis, anti-DNA antibody formation, local infection, and tumor nodule ulceration, restrict its use (56) . The other limiting factors include immunogenicity in ADEPT and difficulties with the selective delivery and expression of genes in GDEPT (5) . Regarding VDEPT, most viral vectors are engineered to be replication deficient. However, there is a slight risk of reversion to wild-type virus. Furthermore, retrovirus vectors are inserted into the host-cell DNA, which may cause mutagenesis of the host’s genome (11) . Another drawback associated with retroviral vectors is that they only target dividing cells (40, 41) . Even in a rapidly growing tumor nodule, only 6–20% of cells are in a proliferating state and in S phase (71) . Thus, the majority of the tumor would not be sensitive to killing mediated by retroviral VDEPT. On the basis of these variables, the choice for GDEPT, VDEPT, or ADEPT should depend on the clinical scenario and is determined by how developers view the risks associated with each approach. Recent clinical trials of enzyme/prodrug therapy are summarized in Table 5 .
There are three major aspects that need to be improved for enzyme/prodrug combination therapy in the future.
Improved Prodrugs.
The design, synthesis, and clinical trials of prodrugs were recently reviewed elsewhere (73) . One limitation for the prodrug/enzyme approach is that only a small part of the tumor cells become activation competent with current strategies. To overcome this problem, the design of appropriate prodrugs that can diffuse efficiently and can kill activation-incompetent cells via a bystander effect is necessary (5) . Because hypoxia and lower pH are a common environmental feature in solid tumors, there is a need to design prodrugs, which can be activated under these conditions (5) . Currently available systems, including HSV-TK/GCV and CD/5-FC, are dependent on ongoing DNA replication in proliferating cells (20) . Because the majority of tumor cells are in a nonproliferating state, these two commonly used systems are not very effective in killing tumor cells. The ideal active drugs should be effective against both dividing and nondividing cells. Unfortunately, most of the prodrugs used now are antimetabolites and target only dividing cells for cytotoxicity (13, 20) . Alkylating agents derived from the prodrugs CB1954 or IFA are not cell phase specific (74) and may represent a prototype for the development of other novel prodrugs in this class. In a series of CB1954 derivatives, evaluated in a Chinese hamster cell line transfected with the E. coli NTR, 4 of 20 analogues were more potent cytotoxic agents than the parent compound (15) . Another novel prodrug amino-seco-CBI-TMI, a potent minor groove alkykating agent, was synthesized and tested in human ovarian carcinoma cells, where this novel drug gave a 10–21-fold increase in cytotoxicity in the presence of E. coli B NTR (74) . However, all of these new prodrugs have the additional hurdle of FDA approval for use in humans, as well as evaluation in combination with enzyme system.
In addition to alkylating agents, other classes of prodrugs have been developed as well. Some effort was made to improve the water solubility, stability in blood, and susceptibility to enzymatic cleavage for camptothecin (1) , an antitumor alkaloid which acts by inhibiting the activity of topoisomerase I (75) . The other efforts were aimed at the synthesis of a series of new prodrugs of daunorubicin and doxorubicin to find a better substrate for enzyme β-glucuronidase (65, 76) .
Improved Enzymes.
The techniques used to improve enzymes to activate prodrugs are reviewed elsewhere (77) . Use of substrates for human enzymes may allow prodrug activation in nontumor tissues. One solution to this problem is to develop a mutant form of human enzymes by site-directed mutagenesis to avoid immune response against nonhuman protein and improve the kinetics of the enzymes for the prodrugs (77, 78) or make the prodrug a highly specific substrate for the enzyme (3, 13) . In addition, because certain prodrugs may be activated by a cascade of several enzymes, the cotransfection of genes for each member of the pathway is an alternative to increase the yield of active drugs (3) . Finally, use of enzymes from different species may provide another way to improve enzymatic activity (13) , e.g. , yeast CD is more efficient at converting 5-FC to 5-FU than bacterial CD after retroviral infection in murine squamous carcinoma cells and in a mice model of squamous cell cancer of the head and neck (42) . However, this does not avoid the concern over immunogenicity of nonhuman proteins.
Improved Methods to Deliver Prodrug-activating Enzymes into Tumor Tissues.
Using current delivery systems, only 10–55% of cells can be targeted, depending on the tumor and the delivery route (61) . The physical/chemical delivery methods, such as electroporation, direct intracellular injection, and calcium phosphate coprecipitation have been successful in vitro , but clinically, they are only suitable for transfection of tissues that can be removed from the body and then easily returned (11) . Therefore, the improvement in design of delivery vectors of therapeutic genes into tumor cells and development of nonviral vectors are expected (43, 79, 80, 81) . Because of the risks of VDEPT and multiple steps involved to generate functional enzymes in GDEPT, developing nonviral vectors that are able to deliver active enzymes rather than genes into tumors are beneficial for clinical application. Because antibody-enzyme conjugates are large molecules that are difficult to penetrate into tumors using ADEPT, developing a novel approach that is quick, efficient, and involves a small molecule as a targeting agent is needed.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Supported by the Siteman Cancer Center.
The abbreviations used are: GDEPT, gene-directed enzyme prodrug therapy; ADEPT, antibody-directed enzyme prodrug therapy; CB1954, 5-(aziridin-1-yl)-2,4-dinitrobenzamide; CD, cytosine deaminase; CEA, carcinoembryonic antigen; CES, carboxylesterase; CMDA, 4-[(2-chloroethyl)(2-mesyloxyethyl) amino] benzoyl- l -glutamic acid; CPA, carboxypeptidase A; CPG2, carboxypeptidase G2; CPT-11, 7-ethyl-10- [4-(1-piperidino)-1-piperidino] carbonyloxy-camptothecin (irinotecan); CYP, cytochrome P450 isozyme; 5-FC, 5-fluorocytosine; 5-FU, 5′-fluorouracil; GCV, ganciclovir; GPAT, genetic prodrug activation therapy; HSV, herpes simplex virus; IFA, ifosfamide; MTX, methotrexate; NTR, nitroreductase; ODC, ornithine decarboxylase; RED, flavoenzyme NADPH-p450 reductase; TK, thymidine kinase; TRE, tumor-specific transcriptional regulatory elements; VDEPT, virus-directed enzyme prodrug therapy.

General outline of all approaches for enzyme/prodrug cancer therapy.

Use of the ODC promoter to drive N-myc-mediated overexpression of a rabbit CES to achieve selective activation to SN-38 in neuroblastoma cells. Modified from Pawlik et al. (59) .
Selected examples of GDEPT
| Enzymes . | Prodrugs . | Model systems . | References . |
|---|---|---|---|
| Human β-glucuronidase | HMR 1826 | Tumor cells and xenograft model in nude mice | 2 |
| Bacterial nitroreductase | CB1954 | Chinese hamster and 3T3 cells | 15, 23 |
| Carboxypeptidase | MTX-α-peptide | Cos-1 cells | 4 |
| CYP2B1 and p450 reductase | Cyclophosphamide | Rat 9L gliosarcoma cells | 18 |
| Rabbit CYP4B1 | 2-AA or 4-IM | Human, rat glioma cells and in nude mice tumor model | 12 |
| Thymidine phosphorylase | 5-FU or 5′-DFUR | LS 174T human colon carcinoma cells | 1 |
| Rabbit and human carboxylesterase | Irinotecan | Glioblastoma and rhabdomyosarcoma cells and preclinical mouse xenograft model | 24 |
| β-galactosidase | Anthracycline | Human melanoma cells | 25 |
| Cytosine deaminase | 5-FC | Murine fibroblast cells | 21 |
| Thymidine kinase | GCV | Cisplatin-resistant human ovarian carcinoma cells | 26 |
| Enzymes . | Prodrugs . | Model systems . | References . |
|---|---|---|---|
| Human β-glucuronidase | HMR 1826 | Tumor cells and xenograft model in nude mice | 2 |
| Bacterial nitroreductase | CB1954 | Chinese hamster and 3T3 cells | 15, 23 |
| Carboxypeptidase | MTX-α-peptide | Cos-1 cells | 4 |
| CYP2B1 and p450 reductase | Cyclophosphamide | Rat 9L gliosarcoma cells | 18 |
| Rabbit CYP4B1 | 2-AA or 4-IM | Human, rat glioma cells and in nude mice tumor model | 12 |
| Thymidine phosphorylase | 5-FU or 5′-DFUR | LS 174T human colon carcinoma cells | 1 |
| Rabbit and human carboxylesterase | Irinotecan | Glioblastoma and rhabdomyosarcoma cells and preclinical mouse xenograft model | 24 |
| β-galactosidase | Anthracycline | Human melanoma cells | 25 |
| Cytosine deaminase | 5-FC | Murine fibroblast cells | 21 |
| Thymidine kinase | GCV | Cisplatin-resistant human ovarian carcinoma cells | 26 |
2-AA, aminoanthracene; 4-IM, 4-ipomennol; D′FUR, 5′-deoxy-5-fluorouridine.
Selected examples of VDEPT
| Viral vectors . | Enzymes delivered . | Prodrugs . | Model system . | References . |
|---|---|---|---|---|
| Adenovirus | Herpes simplex virus thymidine kinase | GCV | Mouse prostate cancer cell line and clinical trials | 19, 37, 38 |
| Human carboxylesterase | Irinotecan | Human lung adenocarcinoma cell lines and nude mice tumor model | 39 | |
| nitroreductase | CB1954 | Ovarian tumor cells and animal model of disseminated intraperitoneal carcinoma | 40, 41 | |
| Retrovirus | nitroreductase | CB1954 | Colorectal, pancreatic, ovarian cancer cells and xenografts of human ovarian & pancreatic cancer | 20, 30, 41 |
| Yeast cytosine deaminase | 5-FC | SCC VII murine squamous carcinoma cells and YCD-expressing tumors | 42 | |
| Human CYP and p450 reductase | Cyclophosphamide and IFA | Gliosarcoma cells and tumor model | 43 | |
| EBV | Nitroreductase | CB1954 | EBV-positive B-cell lines | 35 |
| Viral vectors . | Enzymes delivered . | Prodrugs . | Model system . | References . |
|---|---|---|---|---|
| Adenovirus | Herpes simplex virus thymidine kinase | GCV | Mouse prostate cancer cell line and clinical trials | 19, 37, 38 |
| Human carboxylesterase | Irinotecan | Human lung adenocarcinoma cell lines and nude mice tumor model | 39 | |
| nitroreductase | CB1954 | Ovarian tumor cells and animal model of disseminated intraperitoneal carcinoma | 40, 41 | |
| Retrovirus | nitroreductase | CB1954 | Colorectal, pancreatic, ovarian cancer cells and xenografts of human ovarian & pancreatic cancer | 20, 30, 41 |
| Yeast cytosine deaminase | 5-FC | SCC VII murine squamous carcinoma cells and YCD-expressing tumors | 42 | |
| Human CYP and p450 reductase | Cyclophosphamide and IFA | Gliosarcoma cells and tumor model | 43 | |
| EBV | Nitroreductase | CB1954 | EBV-positive B-cell lines | 35 |
Selected examples of GPATs
| Promoters . | Enzyme genes . | Model system . | References . |
|---|---|---|---|
| Ornithine decarboxylase | Rabbit carboxylesterase | Neuroblastoma cells overexpressing N-myc | 59 |
| Regulatory elements of MUC1 and B2 | Herpes simplex virus thymidine kinase | MUC1-positive cells | 57 |
| CEA tumor antigen | Herpes simplex virus thymidine kinase | CEA-producing gastric cancer cell lines | 11 |
| α-fetoprotein enhancer | Herpes simplex virus thymidine kinase | Hepatocellular carcinoma cell lines | 11 |
| B2 | Cytosine deaminase | Breast and pancreatic tumor cells | 11 |
| Human tyrosine promoter | Purine nucleoside phosphorylase | Melanoma cell lines | 11 |
| Promoters . | Enzyme genes . | Model system . | References . |
|---|---|---|---|
| Ornithine decarboxylase | Rabbit carboxylesterase | Neuroblastoma cells overexpressing N-myc | 59 |
| Regulatory elements of MUC1 and B2 | Herpes simplex virus thymidine kinase | MUC1-positive cells | 57 |
| CEA tumor antigen | Herpes simplex virus thymidine kinase | CEA-producing gastric cancer cell lines | 11 |
| α-fetoprotein enhancer | Herpes simplex virus thymidine kinase | Hepatocellular carcinoma cell lines | 11 |
| B2 | Cytosine deaminase | Breast and pancreatic tumor cells | 11 |
| Human tyrosine promoter | Purine nucleoside phosphorylase | Melanoma cell lines | 11 |
Examples of ADEPT in enzyme/prodrug cancer therapy
| Therapy route . | Enzymes . | Antibodies . | Prodrugs . | Model systems . | References . |
|---|---|---|---|---|---|
| β-glucosidase | Bladder cancer-associated monoclonal antibody | Amygdalin | HT 1376 bladder cancer cells | 64 | |
| Human β-glucuronidase | Humanized CEA-specific binding region | A series of new prodrugs of anthracyclines | Murine L 1210 tumor cell line | 65 | |
| Human β-glucuronidase | Single-chain anti-CD20 antibody | Doxorubicin | Fused protein | 67 | |
| Human β-glucuronidase | Humanised Fab fragments of the anti-CEA Mab | Doxorubicin | Fused protein | 68 | |
| Carboxy-peptidase G2 | Anti-CEA antibody | CMDA | Xenograft of human colon carcinoma | 63 |
| Therapy route . | Enzymes . | Antibodies . | Prodrugs . | Model systems . | References . |
|---|---|---|---|---|---|
| β-glucosidase | Bladder cancer-associated monoclonal antibody | Amygdalin | HT 1376 bladder cancer cells | 64 | |
| Human β-glucuronidase | Humanized CEA-specific binding region | A series of new prodrugs of anthracyclines | Murine L 1210 tumor cell line | 65 | |
| Human β-glucuronidase | Single-chain anti-CD20 antibody | Doxorubicin | Fused protein | 67 | |
| Human β-glucuronidase | Humanised Fab fragments of the anti-CEA Mab | Doxorubicin | Fused protein | 68 | |
| Carboxy-peptidase G2 | Anti-CEA antibody | CMDA | Xenograft of human colon carcinoma | 63 |
Examples of recent clinical trials of enzyme/prodrug therapy Approach
| Approach . | Enzyme . | Prodrug . | Efficiency . | Reference . |
|---|---|---|---|---|
| GPAT | CD cDNA directed by B-2 promoter | 5-FC | Plasmid construct was intratumorally injected in 12 breast cancer patients. | 56 |
| Expression was targeted to B2-positive cells and occurred in 90% of cells. | ||||
| ADEPT | CPG2 linked to F(ab′) fragment of murine A5B7 monoclonal antibody | CMDA | The concentration of active drug (CJS11) in plasma of 10 patients with colorectal carcinoma was evaluated. | 70, 72 |
| On biopsies, CPG2 activity was only localized in metastatic tumor. | ||||
| VDEPT | Adenoviral transduction of HSV-TK | GCV | Recombinant adenovirus containing HSV-TK was injected into the pleural cavity of 21 patients with mesothelioma. HSV-TK was detected in tumors of 11 patients. | 37, 38 |
| Intraprostatic injection of recombinant adenovirus containing HSV-TK was administered to 18 patients with local recurrence of prostate cancer. Fall in serum PSA by 50% was observed for 6 weeks to 1 year in 3 patients. | 45 | |||
| Retroviral transduction of HSV-TK | GCV | Gene therapy in combination with surgery was applied to 48 patients with GBM. No significant side effects were observed, and the 12-month survival rate was 27%. | 44, 464748 |
| Approach . | Enzyme . | Prodrug . | Efficiency . | Reference . |
|---|---|---|---|---|
| GPAT | CD cDNA directed by B-2 promoter | 5-FC | Plasmid construct was intratumorally injected in 12 breast cancer patients. | 56 |
| Expression was targeted to B2-positive cells and occurred in 90% of cells. | ||||
| ADEPT | CPG2 linked to F(ab′) fragment of murine A5B7 monoclonal antibody | CMDA | The concentration of active drug (CJS11) in plasma of 10 patients with colorectal carcinoma was evaluated. | 70, 72 |
| On biopsies, CPG2 activity was only localized in metastatic tumor. | ||||
| VDEPT | Adenoviral transduction of HSV-TK | GCV | Recombinant adenovirus containing HSV-TK was injected into the pleural cavity of 21 patients with mesothelioma. HSV-TK was detected in tumors of 11 patients. | 37, 38 |
| Intraprostatic injection of recombinant adenovirus containing HSV-TK was administered to 18 patients with local recurrence of prostate cancer. Fall in serum PSA by 50% was observed for 6 weeks to 1 year in 3 patients. | 45 | |||
| Retroviral transduction of HSV-TK | GCV | Gene therapy in combination with surgery was applied to 48 patients with GBM. No significant side effects were observed, and the 12-month survival rate was 27%. | 44, 464748 |
PSA, prostate-specific antigen; GBM, glioblastoma multiforme.
Citing articles via
Email alerts.
- Online First
- Collections
- Online ISSN 1557-3265
- Print ISSN 1078-0432
AACR Journals
- Blood Cancer Discovery
- Cancer Discovery
- Cancer Epidemiology, Biomarkers & Prevention
- Cancer Immunology Research
- Cancer Prevention Research
- Cancer Research
- Cancer Research Communications
- Clinical Cancer Research
- Molecular Cancer Research
- Molecular Cancer Therapeutics
- Information on Advertising & Reprints
- Information for Institutions/Librarians
- Privacy Policy
- Copyright © 2023 by the American Association for Cancer Research.
This Feature Is Available To Subscribers Only
Sign In or Create an Account
We use cookies to enhance our website for you. Proceed if you agree to this policy or learn more about it.
- Essay Database >
- Essay Examples >
- Essays Topics >
- Essay on Drugs
Free Classification Of Prodrugs: Essay Sample
Type of paper: Essay
Topic: Drugs , Design , Aliens , Water , Actions , Community , Lipid , Fat
Words: 1200
Published: 02/20/2023
ORDER PAPER LIKE THIS
The prodrug design is an efficient and powerful method that can be practised in a wide range of parent drug compounds, administration routes and preparations. Its main goal is to overcome the drawbacks of regular drug compounds by effectively targeting the drug's particular site of action for improved therapy. This assignment seeks mainly to depict the different classifications of prodrugs and the role these play in drug design through the use of several examples. A brief description highlighting the modern aspects of prodrug design is also covered here.
Introduction:
Prodrugs are inactive forms of drug compounds that undergo chemical or enzymatic conversion inside the body to form an active drug compound. The purpose of a prodrug design is to improve the distribution of that particular drug in systemic circulation by either increasing or decreasing its aqueous and lipid solubility. Prodrugs have also been reported to improve chemical stability and reduce toxicity (Patrick, 2013). Studies have found that 10% of the drugs marketed around the world currently fall under the prodrugs category. In 2008 alone, 33% of all the totally authorized minute-molecular-weight drugs were prodrugs. (Huttunen et al; 2011)
Role of prodrugs in drug design:
Imroving and reducing water solubility Aqueous solubility can be increased by attaching the polar neutral groups like amino acids, sugar molecules or phosphates to the parent drug (Zawilska et al; 2013). This approach is usually employed in parentral prepration in order to avoid pain at the site of injection. Given that Prednisolone on its own is less soluble in water, attaching the phosphate molecule to the free hydroxyl group of prednisolone renders it more water-soluble and hence increases the drug's effectiveness. Drugs that have unpleasant tastes can prove difficult to swallow as they dissolve on the tongue. This problem can be addressed by decreasing the water solubility of the drug. Chloramphenicol, for example, tastes bitter when consumed orally. Therefore, the drug is combined with palmatic acid to form chloramphenicol palmitate, which is less soluble in water than chloramphenicol. As the palmitate ester does not dissolve on the tongue, it makes the patient more accepting of the medication (Silverman, 2012).
Improving lipid solubility
Drugs that have poor lipid solubility have been reported to lead to poor membrane permeability. This condition can be improved by converting more water soluble molecules - such as phosphate, hyroxyl, thiol, carboxyl or amine - from the parent group to more lipid-soluble groups - like alkyl or aryl ester - which can cross the fatty cell membrane with greater ease. This prodrug approach is most commonly used for oral, topical, transdermal and occular administration. Oseltamivir prodrug, which is used for the treatment of Influenza A and B virus, undergoes hydrolysis to form the active form of oseltamivir carboxylate. Converting the hydroxyl group to ester creates oseltamivir lipophilic, which can cross the cell membrane with far greater ease (Huttunen et al; 2011).
Enhancing drug stability
When administered orally, most drugs are metabolized, which serves to reduce the actual amount of the drug that reaches the target. The development of prodrug design seeks to solve this problem by attaching the carrier group to the metabolically-liable group of the parent drug thereby preventing the breakdown of drug until it reaches its intended target. Bambuterol, for example, is a prodrug of terbutaline. It is used in asthma treatment. Terbutaline contains two metabolically liable phenol groups that lower the bioavailability of the drug. This is improved by converting the phenolic groups to N,N-dimethyl carbamate ester to obtain bambuterol, which is far more stable to hydrolysis (Huttunen et al; 2011).
Reducing toxic and side effects
In order to best attain the therapeutic benefits of an active drug , its toxicity and side effects should always be kept to a minimum level. Doxorubicin, an anti-cancer drug, has been proven to cause cardiotoxicity. To overcome this particular side effect, a galactoside prodrug was attached to doxorubucin through the use of a carbamate spacer, which simultaneously increases the doxorubicin availability in tumor tissue and decreases its accumulation in the cardiac tissue. Moreover, the hydrophilic nature of galactoside allows it to avoid further distribution to other tissues, rendering it less toxic and more effective (Stella et al; 2007).
Increasing the duration of action
Drugs with lower half-lives require repeated dosing in order to retain blood concentration, which can leas to poor patient acceptability over time. This is solved by increasing the duration of action. Fluphenazine deconate, which is the prodrug of fluphenazine, has long been used as a long acting IM injection to treat Schizophrenia. This injection, given once every two weeks, has been shown to increase both its effectiveness and patient acceptability (Patrick, 2013).
Regular functional groups used in prodrug design
Modern aspects of prodrug design: In the past, the approach to prodrug design focused on altering the different physiochemical conditions such as solubility, absorption , permeability, distribution etc. However, these approaches lacked site specificity. The modern prodrug approach, by contrast, is based on molecular and cellular factors (Dahan et al; 2014). Here, the carrier molecule attach covalently to the molecule to selectively target enzymes or transporters . This innovative approach provides a far more significant prospective for enhancing drug bioavailability and choosiness amongst poorly absorbed drug molecules.
Targeting transpoters
Targeting enzymes Enzymes are employed to increase both overall oral drug absorption and site-specific drug delivery. The use of a nutrient molecule can help provide more specific targeting in order to enhance oral absorption. In site-specific drug delivery, the active drug is released only at its intended site of action, thereby reducing the toxic effect that might be caused due to non-specific uptake by other tissues. This approach is usually used for cancer chemotherapy, in which a non-toxic drug is converted into a cytotoxic by an enzyme at the tumour site (ADEPT) or in neoplastic cells (GDEPT) (Dahan et al; 2014).
Conclusion:
Progress in molecular biology has led to equally significant changes to the pharmaceutical sciences domain. In earlier times, the prodrug methodology was only considered as a last resort option, only to be used when experts had attempted all other possible options first. Nowadays, the prodrug approach is applied at the very early stages of the development process and is now considered a more powerful and successful alternative.
Works Cited
Patrick, G. L. (2013) An introduction to Medicinal Chemistry, 5thedn. Oxford: Oxford University Press. Thomas, G. (2003) Fundamentals of medicinal chemistry, West Sussex: John Wiley & Sons Ltd. Silverman, Richard, B. (2012) The organic chemistry of drug design and drug action, Elsevier Science. Dahan, A., Zimmermann, M. &Ben-Shabat, S. (2014) ‘Modern prodrug design for targeted oral drug delivery’, Molecules19, 1420-3049: 16489-16505. Zawilska, J., Wojcieszak, J. &Olejniczak, B. (2013) ‘Prodrugs: A challenge for the drug development’, Pharmacological Reports 65, 1734-1140: 1-14. Huttunen, M., Raunio, H. &Rautio, J. (2011) ‘Prodrugs- from Serendipity to Rational Design’, Pharmacological reviews 63, 3: 750-771. Stella, V., Borchardt, R., Hageman, M.,Oliyai, R.,Maag, H., Tilley, J. (2007) Prodrugs: Challenges and Rewards,Springer Science & Business Media

Cite this page
Share with friends using:
Removal Request

Finished papers: 1513
This paper is created by writer with
ID 253294903
If you want your paper to be:
Well-researched, fact-checked, and accurate
Original, fresh, based on current data
Eloquently written and immaculately formatted
275 words = 1 page double-spaced

Get your papers done by pros!
Other Pages
Priesthood profession personal statement, literary position paper literature review example, a story of modern war book review samples, cicis pizza essay samples, sample course work on protecting food safety, excessive use of cell phone and its negative effect on teenagers health research papers examples, sample essay on my trip to erie metroparks, free essay about plural wives an lds policy at odds with american society, essay on comparison of quot the raven quot by edgar allen poe and the movie quot the raven, good example of term paper on overall theme of the book, free essay about how competitive game industry contributes to economy, good critical thinking about rebecca morris artist painter lecturer, feminist imagery of yahweh in the bible essay example, women and standpoint theory essay example, what are the implications of global warming on irans industry essay example, free impersonal policing vs community policing argumentative essay example, music history essays example, sample essay on recommendation 3, good the rise and fall of the occupy movement research paper example, good example of other theology essay, the fbi and cia in the intelligence framework critical thinkings example, good essay on lesson plan, netiquette essays, internet censorship essays, virulent essays, maca essays, psychoanalytic theory essays, musculoskeletal disorders essays, job market essays, encyclopedia britannica essays, democratic government essays, distribution of wealth essays, what can be done essays, microsoft outlook essays, comical essays, world population essays, marilyn essays, great britain research proposals, balanced scorecard research proposals, legacy research proposals, toxicity research proposals, pledge of allegiance research proposals.
Password recovery email has been sent to [email protected]
Use your new password to log in
You are not register!
By clicking Register, you agree to our Terms of Service and that you have read our Privacy Policy .
Now you can download documents directly to your device!
Check your email! An email with your password has already been sent to you! Now you can download documents directly to your device.
or Use the QR code to Save this Paper to Your Phone
The sample is NOT original!
Short on a deadline?
Don't waste time. Get help with 11% off using code - GETWOWED
No, thanks! I'm fine with missing my deadline
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Transl Clin Pharmacol
- v.26(1); 2018 Mar
Understanding the pharmacokinetics of prodrug and metabolite
Seungil cho.
Molecular Diagnostics and Imaging Center, School of Medicine, Kyungpook National University; Clinical Trial Center, Kyungpook National University Hospital, Daegu 41944, Korea.
Young-Ran Yoon
This tutorial explains the pharmacokinetics of a prodrug and its active metabolite (or parent drug) using a two-step, consecutive, first-order irreversible reaction as a basic model for prodrug metabolism. In this model, the prodrug is metabolized and produces the parent drug, which is subsequently eliminated. The mathematical expressions for pharmacokinetic parameters were derived step by step. In addition, we visualized these expressions to help understand the relationship between pharmacokinetic parameters easily. For the elimination rate-limited and formation rate-limited metabolism, we analyzed the plasma drug concentration versus time curve of a prodrug administered intravenously.
Introduction
Prodrug is a pharmacologically inactive derivative of an active drug and undergoes in vivo biotransformation to release the active drug by chemical or enzymatic cleavages.[ 1 , 2 , 3 ] A prodrug strategy is typically used when a pharmacologically active drug has poor solubility or permeability.[ 1 , 2 ] Various chemically or enzymatically labile functional groups have been introduced to improve the properties of the parent drug and decrease the presystemic metabolism.[ 1 , 2 ] Many successful examples have been reviewed in the literature.[ 1 , 2 , 3 ] A detailed discussion of prodrugs and their active metabolites (parent drugs) is beyond the scope of this tutorial. Here we focus on the pharmacokinetics of a prodrug and its metabolite to help understand their pharmacokinetic data obtained from preclinical and clinical studies. This will ultimately help define the proper dose of the prodrug needed for efficacy.
Theoretical Analysis
Prodrug kinetics.
When a prodrug is administered orally, it undergoes complicate processes of absorption, distribution, metabolism, and excretion. To understand these processes for metabolites, Cummings and Martin published results on the excretion and accrual of drug metabolites in 1963.[ 4 ] This provides the basis for the pharmacokinetics of drug and its metabolites and can be readily applied to study the pharmacokinetics of prodrug. Since then, many theoretical analyses and reviews, related to prodrug pharmacokinetics, have been published.[ 5 , 6 , 7 , 8 , 9 , 10 ] A thorough understanding of prodrug kinetics is daunting and may be achieved using sophisticated physiologically based pharmacokinetic (PBPK) modeling.[ 11 , 12 ]
To get some insight pertinent to prodrug kinetics, we consider a simple scheme for the fate of a prodrug (P) after intravenous administration ( Fig. 1 ). A fraction of the prodrug ( f ) is metabolized and produces a parent drug (D) with a first-order formation rate constant, k f(D) . The drug (D) may be further metabolized to a daughter metabolite (DM 1 ) or excreted. The elimination rate constant of the parent drug, k el(D) , is expressed by the sum of the formation rate constant of the daughter metabolite, k f(DM1) , and the urinary excretion rate constant, k ex(D) . More than one metabolite may be formed with a formation rate constant, k f(PM2) , and then excreted with a rate constant, k ex(PM2) . Otherwise, a fraction of the drug is excreted into urine as unchanged form (P) with a rate constant, k ex(P) . The overall elimination rate constant of the prodrug, k el(P) , is the sum of k f(D) , k f(PM2) , and k ex(P) . No matter how complex the pathways may appear, the time course for the change of metabolite amount can be described by a general relationship: rate of change of metabolite amount in body = rate of formation − rate of elimination. [ 5 , 6 ] However, a full mathematical description of this model is still complex and difficult to solve. We will further simplify this model to have clear understanding of the factors influencing the amount of parent drug in the body.

A basic model for prodrug kinetics
Consider a simple case of a two-step consecutive first-order irreversible reaction:
This model provides the basis for developing more complex models. Thus it is crucial to understand this reaction kinetics thoroughly for further study. Here, we use simple notations for the sake of clarity. We can set up the following three differential equations to describe the change in amounts of A, B, and C over time.
At t = 0, A = A 0 and B = C = 0, and A + B + C = A 0 at all times. The Eq. (1) can be solved immediately to give
Substituting Eq. (4) into Eq. (2), we get
Eq. (5) can be solved simply by noting that
By multiplying both sides of Eq. (5) by e k 2 t and using the above relationship, we get
If k 1 ≠ k 2 , integration of Eq. (6) from t = 0 to t gives
In the special case when k 1 = k 2 , Eq. (6) can reduce to
Then, the integrated expression becomes
The expression for C can be obtained using the mass balance relationship, C = A 0 − A − B ,
Figure 2 shows a typical amount-time profile of each species for the consecutive first-order irreversible reaction. The amount of A decreases at the normal exponential rate characteristic of a first-order decay curve (blue line). The amount of B follows a bi-exponential profile, first rising and then declining (red line). The amount of C continuously increases and reaches a plateau (green line). This profile is useful for quantifying absorption and elimination processes of drug as well as formation and elimination processes of metabolite.[ 13 , 14 ]

To calculate the maximum amount of B ( B max ) and the time to reach the peak ( t max ), we take the derivative of Eq. (7), set the derivative, dB/dt , to zero, and rearrange to obtain
Take the natural logarithm of both sides and solve for t max to get
The resulting B max at t max then becomes
Using Eq. (10), we obtain
From Eq. (11), we get
In Figure 2 , we also visualized the Equations (11), (13), and (14) to show that t max decreases and B max increases as the ratio of k 1 to k 2 increases (dashed line). In other words, when k 2 is smaller than k 1 , the amount of A quickly decreases while the amount of B increases rapidly, reaches a maximum at a shorter time, and then falls off with time.
Plasma concentration profile after intravenous administration of prodrug
Now consider a case for the intravenous administration of prodrugs ( P ) in which all the prodrugs are converted to active drugs ( D ) and then eliminated. In this case, the first-order formation rate constant for D , k f(D) , is equal to the elimination rate constant for P , k el(P) . The parameter k el(D) represents the first order elimination rate constant for D . The rate of change of D in the blood is determined by the difference between the formation and the elimination rates. Because the level of each species in the blood is measured in concentration ([ D ] = D /V D , where V D is the volume of distribution for D), it is convenient to express Eq. (7) with the same unit. Then, using new notations ( k 1 → k f(D) ; k 2 → k el(D) ), we get
where V P is the volume of distribution for P, and [P] 0 is the initial concentration of P in the blood.
When plasma concentrations are measured as a function of time, the area under the concentration-time curve (AUC) is of great interest in pharmacokinetics. We can derive a useful relationship between AUC and clearance (CL = k ×V; volume/time in unit) by solving Eq. (2) using a concentration-time integral method. Writing Eq. (2) in new notations and taking the integral of both sides, we get
Because D is not present in the body initially or at infinity, the left side becomes zero. Rearranging Eq. (16) and substituting k el(P) = CL (P) /V P and k el(D) = CL (D) /V D , we get
This ratio is useful to study metabolic pathway and pharmacokinetic interactions.[ 8 , 9 , 10 , 13 ]
Elimination rate-limited (ERL) and formation rate-limited (FRL) metabolism
As shown in Eq. (15), the plasma drug concentration, [ D ], at any given time is expressed by two exponential functions. Depending on the relationship between the prodrug and drug elimination rates, one term can become more dominant than the other. For a more detailed discussion, we consider two limiting situations. In the first situation, formation is a much faster process than elimination (ERL metabolism: k f(D) > k el(D) ). In the second situation, formation proceeds much more slowly than elimination (FRL metabolism: k f(D) < k el(D) ). To help understand the underlying kinetics, we simulated these situations to draw semi-logarithmic plots of plasma concentration versus time using Eq. (15) ( Fig. 3 ).

In ERL metabolism, at large time, the first term approaches zero because k f(D) > k el(D) , and Eq. (15) reduces to
Taking the logarithm to the base 10 of Eq. (18), we get
Thus, the semi-logarithmic plots of plasma concentration versus time become linear with a slope of k e l D 2.303 at large time. Back extrapolation (dashed line) of the elimination phase slope (red line) provides an estimate of [D] 0 , which is the intercept of k f D V P P 0 V D k f D − k e l D ( Fig. 3a ). When k f(D) >> k el(D) and V P = V D , the intercept can be further reduced to [P] 0 .
This situation is commonly encountered in the metabolism of many prodrugs.[ 10 , 15 ] An example is the metabolism of an antitrypanosomal compound (OSU-36) after an intravenous administration of its ester prodrug (OSU-40) to rat. The hydrolysis of OSU-40 was fast, and its concentrations declined rapidly with short half-life (4.8 min). Whereas, the concentrations of OSU-36, hydrolyzed from OSU-40, declined slowly with the half-life of 41.9 min. This was close to the elimination half-life of the preformed OSU-36 (43.9 min), directly administered to the systemic circulation by an intravenous injection.[ 15 ]
In FRL metabolism ( k f(D) < k el(D) ), at large time, Eq. (15) can be reduced in a similar way to give
The slope and intercept of the semi-logarithmic plot of plasma concentration versus time are k f D 2.303 and k f D V P P 0 V D k e l D − k f D , respectively ( Fig. 3b ). In the elimination phase, thus, the concentration of the parent drug is governed by the prodrug formation rate, not by the prodrug elimination rate. Since k f(D) < k el(D) , the intercept can be further reduced to
The plasma prodrug concentration, [P], at any given time is easily obtained from Eq. (4), and its final semi-logarithmic expression is
In equations (21) and (23), it is worthwhile to note that the slopes are the same, that is, the concentrations of the parent drug and prodrug decline in parallel ( Fig. 3b ). Because the parent drug is eliminated almost as soon as it is formed, the elimination rate is approximately equal to the formation rate. The above equation can also be used to plot the plasma concentration-time profile of a preformed D (D*) directly administered to the systemic circulation by an intravenous injection (dotted line). From Figure 3b , we can easily figure out that the half-life of the formed parent drug (ln 2/ k f(D) ) in the elimination phase is longer than that of the preformed parent drug (ln 2/ k el(D*) ). The half-life of the formed parent drug reflects that of the prodrug (ln 2/ k el(P) = ln 2/ k f(D) ).
The FRL metabolism is less encountered in prodrug kinetics but occasionally in metabolite kinetics.[ 10 , 13 , 16 ] One interesting example is the metabolism of 3′-azido-2′,3′-dideoxy-5′-O-oxalatoylthymidine (AZT-Ac) to zidovudine, [3′-azido-2′,3′-dideoxythymidine (AZT)].[ 16 ] Only AZT was detected in plasma, indicating that the prodrug is rapidly hydrolyzed in vivo with a high elimination rate constant. Thus, the first step looks like proceeding according to ERL-metabolism. However, the concentrations of AZT metabolized from AZT-Ac (t 1/2 = 2.16 h) declined more slowly than those of AZT from preformed AZT (t 1/2 = 0.96 h). This means that the metabolic pathway proceeds according to FRL metabolism ( k f(AZT) < k el(AZT) ), as discussed above. This discrepancy may be explained by the presence of multiple pathways for AZT-Ac elimination (small f in Fig. 1 ) or by the presence of another intermediate between AZT-Ac and AZT. Because AUC for AZT metabolized from AZT-Ac is slightly larger than that for AZT from preformed AZT at the same intravenous dose, the latter explanation looks more plausible.
In FRL metabolism, it is interesting to consider one more situation where V P / V D > k el(D) / k f(D) > 1, or CL (P) > CL (D) . As shown in Figure 3c , the intercept for the parent drug ([D] 0 ) is greater than that for the prodrug ([P] 0 ). This situation is encountered in the metabolic conversion of propranolol to naphthoxylactic acid (NLA) after single intravenous and oral doses.[ 17 ] The AUC of NLA was two times greater than that of propranolol after an intravenous dose of propranolol (4 mg) and ten times after a single oral dose (20 or 80 mg). This can be easily explained if the volume of distribution of NLA is much smaller than that of propranolol. The volumes of distribution of basic drugs are often larger than 100 L, while those of their acidic metabolites are close to 10~20 L.[ 13 ] Thus, this situation is expected to be common when a basic prodrug is converted to an acidic parent drug.
We described the prodrug kinetics for the simplest situation: formation and sequential elimination. We can consider a more complicated model to account for more realistic situations but will lose simplicity by including more terms in equations. A more detailed discussion is beyond the scope of this tutorial. For further study, we recommend to read articles and book chapters in the References section.
Acknowledgements
This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C2750).
Reviewer: This article was invited and reviewed by the editors of TCP.
Conflicts of interests: -Authors: The authors have no conflicts of interest to declare.
-Reviewers: Nothing to declare
-Editors: Nothing to declare
COMMENTS
Prodrugs can be developed for many routes of administration: oral, intravenous, intramuscular, inhalation, transdermal, and more . Nevertheless, the oral route is the most common and desirable route of administration; therefore, oral prodrugs are emphasized in this article. Our main goals are to provide the key concepts of the prodrug approach ...
Prodrugs have been extensively reviewed from various perspectives 1,2,3,9,15,16,17,18.A previous prodrug Review was published in this journal 10 years ago, in 2008 (Ref. 10).In the present ...
1. Introduction. Prodrugs are biologically inactive compounds that are activated post-administration to their pharmacologically active forms. Often prodrugs are formulated to overcome pharmacokinetic barriers such as poor solubility and absorption, extensive first-pass metabolism, or rapid excretion, and pharmacodynamic barriers such as toxicity, side effects, and poor efficacy.
2. Ester Prodrugs. The features of an ideal prodrug include the following: (a) hydrolysis resistance during absorption; (b) weak or no activity; (c) aqueous solubility; (d) good permeability through the cells; (e) chemical stability at different pHs; (f) kinetics that allow release of the parental drug [].Among the chemical bonds used to link the parental drug and carrier, esters have proven ...
Additionally, only 43% (35 prodrugs) exhibit more moderate Vd ss (20-100 l) whereas more than a quarter (38%, 31 prodrugs) exhibit a high Vd ss (>100 l). As such, prodrugs tend to have a higher ...
The development of prodrugs — chemically modified versions of pharmacologically active agents that must undergo transformationin vivoto release the active drug — is a well-established strategy ...
Prodrugs are bioreversible derivatives of drug molecules that undergo an enzymatic and/or chemical transformation in vivo to release the active parent drug, which can then exert the desired pharmacological effect. In both drug discovery and development, prodrugs have become an established tool for improving physicochemical, biopharmaceutical or ...
Prodrugs are considered to be the bio-reversible or chemical derivatives of drug molecules [2].They need to undergo various enzymatic or chemical modification in vivo to initiate the required pharmacological effect (Fig. 1) [3].Since the late nineteenth century, the concept of prodrug has been utilized to alter the undesirable properties of different drug molecules [4].
Prodrugs are inactive precursors of an active drug designed to be bioconverted (activated) post administration with the main aim of improving the pharmacokinetic properties of the parent drug. Prodrugs have been successful for a longtime. Sulfasalazine, one of the earliest prodrugs, reaches the colon and is metabolized by bacteria into two ...
Drug-metabolizing enzymes and fate of prodrugs: From function to regulation. Tauqeer Hussain Mallhi, ... Yusra Habib Khan, in Biochemistry of Drug Metabolizing Enzymes, 2022. 1 Introduction. Prodrug is a compound with little or no pharmacological activity. It is a precursor compound that metabolizes in the body and converts into a pharmacologically active drug compound.
Bioconversion mechanism for approximately half of the marketed prodrugs relies on the hydrolysis of an ester bond by esterases (Scheme 2) [27].Two equally important reasons for this are i) abundance of hydroxyls and carboxylic acid functionalities in the marketed drugs, and ii) esterases being ubiquitous such that metabolic regeneration of drugs in the body is a facile process [28].
Prodrugs are cunning derivatives of therapeutic agents designed to improve the pharmacokinetics profile of the drug. Within a prodrug, pharmacological activity of the drug is masked and is recovered within the human body upon bioconversion of the prodrug, a process that is typically mediated by enzymes. This concept is highly successful and a ...
Prodrugs are substances administered in an inactive form that is then metabolized in the body in vivo into the active compound. The rationale behind administering prodrugs is to optimize absorption, distribution, metabolism, and excretion of these drugs. Since first described in the 1950s, prodrugs continue to be a fertile area of research.
To accomplish this, retrosynthetic prodrug design highlights the potential of naphthylcombretastatin-based prodrugs, which form highly active cytostatic agents via sequential ring-closing ...
ABSTRACT. Introduction: Prodrugs have been used to improve the selectivity and efficacy of cancer therapy by targeting unique abnormal markers that are overexpressed by cancer cells and are absent in normal tissues.In this context, different strategies have been exploited and new ones are being developed each year. Areas covered: In this review, an integrated view of the potential use of ...
The design and the synthesis of prodrugs for nonsteroidal anti-inflammatory drugs (NSAIDs) have been given much attention by medicinal chemists, especially in the last decade. As a therapeutic group, NSAIDs are among the most widely used prescribed and over the counter (OTC) medications. The rich literature about potential NSAID prodrugs clearly shows a shift from alkyl, aryalkyl or aryl ...
Prodrug. A prodrug is a pharmacologically inactive medication or compound that, after intake, is metabolized (i.e., converted within the body) into a pharmacologically active drug. [1] [2] Instead of administering a drug directly, a corresponding prodrug can be used to improve how the drug is absorbed, distributed, metabolized, and excreted ...
A New Classification of Prodrugs. The primary goal in pharmaceutical design of a prodrug has been to circumvent some disadvantageous pharmacodynamic or pharmacokinetic property of the active drug; e.g., to increase bioavailability or to reduce adverse effects.However, principal concerns during prodrug product development are two-fold: (1) whether the prodrug converts sufficiently fast and ...
The biostability of prodrugs also aims at reducing presystemic metabolism, to enhance time rundown, elevating organ or tissue-selective supply of the active compound. The biostability of prodrugs is influenced by the ease with which enzymes act on the prodrug to liberate the active compound. For example, the esterification of prodrugs with ...
Among all 1,799 approved small-molecule drugs identified from DrugBank (Box 3), only 296 (16%) are anti-infectives and only 202 (11%) are chemotherapeutics. This means that prodrugs are enriched ...
Abstract. The selective activation of prodrug(s) in tumor tissues by exogenous enzyme(s) for cancer therapy can be accomplished by several ways, including gene-directed enzyme prodrug therapy (GDEPT), virus-directed enzyme prodrug therapy (VDEPT), and antibody-directed enzyme prodrug therapy (ADEPT). The central part of enzyme/prodrug cancer therapy is to deliver drug-activating enzyme gene or ...
Introduction: Prodrugs are inactive forms of drug compounds that undergo chemical or enzymatic conversion inside the body to form an active drug compound. The purpose of a prodrug design is to improve the distribution of that particular drug in systemic circulation by either increasing or decreasing its aqueous and lipid solubility.
This situation is commonly encountered in the metabolism of many prodrugs.[10,15] An example is the metabolism of an antitrypanosomal compound (OSU-36) after an intravenous administration of its ester prodrug (OSU-40) to rat. The hydrolysis of OSU-40 was fast, and its concentrations declined rapidly with short half-life (4.8 min). Whereas, the ...